

 POLSKI
POLSKI







Introduction
Primary hyperparathyroidism (PHPT) is a disease that is often detected incidentally during routine biochemical screening in an asymptomatic state [1]. With the development of automated analysers in the 1970s and routine calcium screening, the disease is diagnosed quite early. Asymptomatic state and mild hypercalcaemia are commonly encountered in resource-rich countries [1]. However, in developing countries, the disease continues to manifest as a multi-system disorder with extensive skeletal and renal involvement [2]. Nonetheless, recently, in India also, we have seen a trend towards a less severe state of PHPT [3].
Here, we describe the case of a 16-year-old adolescent girl who had been symptomatic for the past 2 years. She had presented with severe form of the disease with significant emaciation, severe musculoskeletal involvement, renal involvement, and gastro-intestinal manifestation. It is rare to see such severe manifestations of the disease nowadays. We initially suspected parathyroid carcinoma based on her clinical, biochemical, and radiological presentation. However, the final histopathology was suggestive of atypical parathyroid adenoma. Atypical parathyroid adenoma is a relatively rare cause of primary hyperparathyroidism. It falls in the spectrum between adenoma and carcinoma. While the tumour did exhibit a few high-risk characteristics, there was no obvious sign of malignancy.
Case report
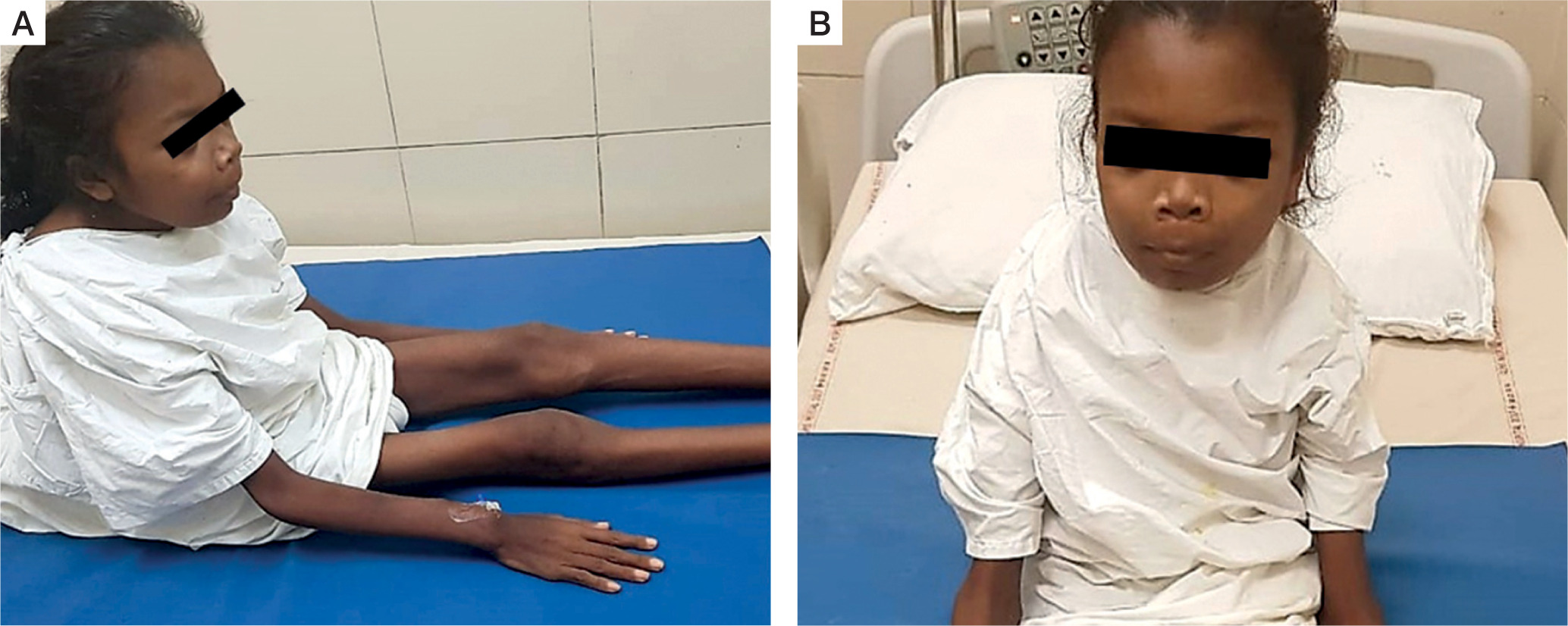
A 16-year-old adolescent girl presented in the endocrine clinic with symptoms of diffuse musculoskeletal pain for 2 years. The intensity of the pain had gradually worsened. Simultaneously, she had generalized weakness involving proximal muscles more than distal. She had difficulty in climbing stairs and getting up from a squatting position. This weakness gradually progressed, and she had become bed bound for the past year. There was no history of overt fracture. For the past 6 years the patient had lost significant weight and was emaciated (Fig. 1A, B). She failed to gain height and remained the shortest among her peers.
She had history of frequent graveluria and flank pain, right more than left. However, she had not undergone any investigation or intervention for renal stones prior to presentation.
Figure 1
Clinical images of the patient: A) patient is bed-ridden with extreme emaciation, dorsal kyphosis of spine; B) clinical image showing frontal prominence and mild prominence of jaws
She gave a history of recurrent reflux symptoms and had to take over-the-counter proton pump inhibitors for dyspeptic symptoms. There was no history of recurrent vomiting or haematemesis. The patient had a loss of appetite over the years along with a history of chronic constipation. Three months prior to the current presentation, she was diagnosed with an episode of acute pancreatitis during evaluation of abdominal pain. Pancreatitis was managed conservatively without any residual complication.
There was no history of hypertension, adrenergic spells, goitre, galactorrhoea, neuroglycopaenic symptoms, polyuria, or behavioural abnormalities. There was no family history of similar illness or any bone-related disease. The patient had not attained menarche until 16 years of age. Tanner pubertal staging revealed a pubertal state of B2 (breast budding) and P2 (scanty pubic hair along the labia). This suggested that the patient had entered puberty but subsequently had pubertal arrest owing to chronic systemic illness.
Physical examination revealed an emaciated state with a height of 140 cm, body weight of 25 kg, and body mass index of 12.1 kg/m2. Her blood pressure in the right arm in a supine position was 90/60 mm Hg, which was within the normal centiles. Height corresponded to –3.02 standard deviation score (SDS) below the mean height for age and sex. Weight corresponded to –2.63 SDS below the mean weight for age and sex. Both height and weight were plotted on an Indian Academy of Paediatrics growth chart for adolescent girls aged 5–18 years. Mid-arm circumference was 14.5 cm on the right arm and 14.0 cm on the left arm, measured with a non-stretchable tape. There was diffuse musculoskeletal tenderness. There was facial dysmorphism with frontal bossing and prominence of the jaws (Fig. 1A). There was no bone swelling on oral examination. There was no palpable neck mass. There was thoracic kyphoscoliosis with convexity to the right but no genu varum, genu valgum, windswept deformity, or contracture deformity of upper or lower limbs. Neurological examination revealed power of 3/5 in the proximal muscles and 4/5 in the distal muscles of the upper and lower limbs.
After admission in our ward, the patient was evaluated and found to have severe PTH dependent hypercalcemia. Her serum total calcium (albumin corrected calcium) was very high, at 18.5 mg/dl (8.5–10.5 mg/dl), and serum inorganic phosphorus was low, at 1.9 mg/dl (2.5–5.0 mg/dl). Serum alkaline phosphatase (ALP) was very high, at 2015 IU/l (80–240 IU/l) along with very high serum intact parathyroid hormone (iPTH) > 5000 pg/ml, (15–65 pg/ml) and vitamin D deficiency, with serum 25(OH)D of 19.1 ng/ml (30–100 ng/ml). She had hypercalciuria with 24-hour urinary calcium of 264 mg/day (> 4 mg/kg body weight). She had deranged renal function tests with serum creatinine of 1.1 mg/dl, and an estimated glomerular filtration rate of 52.2 ml/min/1.73 m2 (Schwartz formula for calculation of GFR in the paediatric age group). She had hypoproteinaemia with serum total protein of 5.8 g/dl (6–8 g/dl) and hypoalbuminaemia with serum albumin of 2.3 g/dl (3–5 g/dl). She also had normocytic normochromic anaemia, with haemoglobin of 8.1 g/dl (11–13 g/dl). Thyroid function tests were suggestive of sick euthyroid syndrome: serum thyroxine (T4) of 4.8 ug/dl (5.1–14.1 ug/dl) and serum thyroid stimulating hormone (TSH) of 0.086 uIU/ml (0.4–4.2 uIU/ml). Serum prolactin (PRL) was normal at 13.1 ng/ml (5–25). Serum gastrin levels were not measured because hypercalcaemia is a known stimulator of gastrin levels.
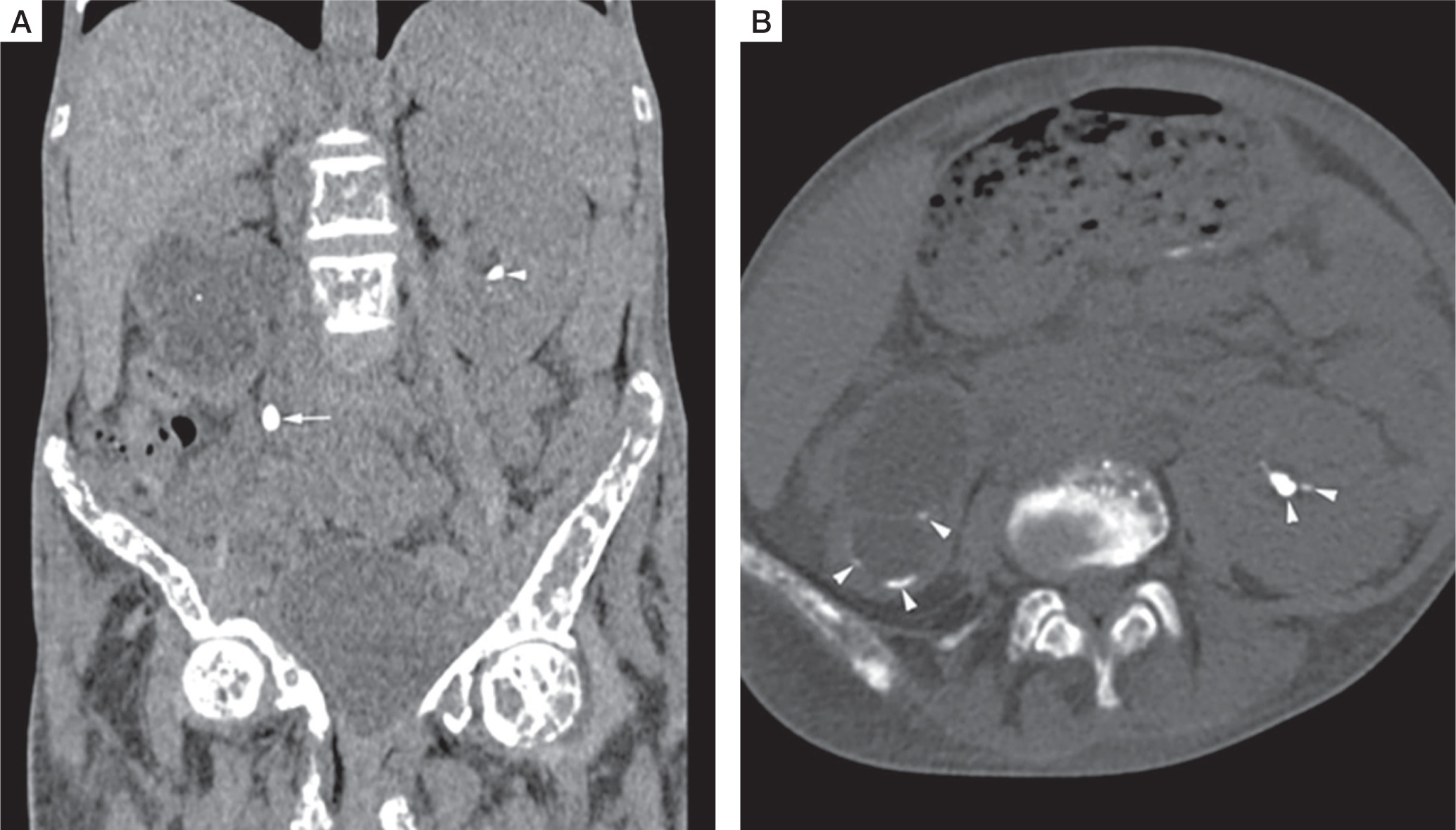
A skeletal survey revealed classical manifestations of PHPT. She had diffuse osteopaenia with multiple brown tumours (osteitis fibrosa cystica). Radiographs of the hands revealed subperiosteal resorption of the digits, prominent on the radial aspects, along with intra-cortical tunnelling (Fig. 2A). A lateral radiograph of the spine showed multiple vertebral crush fractures (Fig. 2B). A skull radiograph revealed multiple, small, well-defined lucencies in the calvaria, giving an appearance of ‘pepper pot skull’ (Fig. 2C). Pelvic radiograph showed triradiate pelvis, with pseudo-widening of the sacroiliac joints and subchondral resorption of the sacroiliac joints and pubic symphysis (Fig. 2D). Posteroanterior radiograph of the chest (Fig. 2E) showed scoliosis with convexity to the right, and multiple brown tumours in the bilateral humeri and right scapula (blue arrowheads). Features of demineralization were noted in all the radiographs. Ultrasound of the kidney showed multiple renal stones. Non-contrast computed tomography (NCCT) of the kidney, ureter, and urinary bladder (KUB) showed right ureteric calculus causing right hydroureteronephrosis (Fig. 3A, B) and bilateral renal calculi. Renal dynamic scan showed left kidney function of 93%, while right kidney function was only 7%.
Figure 2
Skeletal survey: Diffuse osteopenia with coarse trabecular pattern seen. Anteroposterior radiograph of the left hand (A) shows subperiosteal resorption, predominantly along radial aspect of proximal and middle phalanx of 2nd and 3rd finger (white arrowheads),cigar/oval shaped intracortical resorption (white arrows), and subchondral resorption at ulnar side of distal radioulnar joint (black arrow). Lateral radiograph of spine (B) shows biconcave/crush fracture of multiple vertebral bodies with thoracic kyphosis. Lateral radiograph of skull (C) shows multiple small well defined lucencies due to trabecular bone resorption, giving an appearance of ‘pepper pot skull’. Anteroposterior radiograph of the pelvis (D) shows protrusio acetabuli on the right side (black arrowhead), with subchondral resorption at symphysis pubis, and bilateral sacroiliac joints (black asterisks).Healed fracture of right superior and inferior pubic rami are also noted. Posteroanterior radiograph of chest (E) shows scoliosis with convexity to right, and multiple brown tumors in bilateral humerus and right scapula (blue arrowheads)
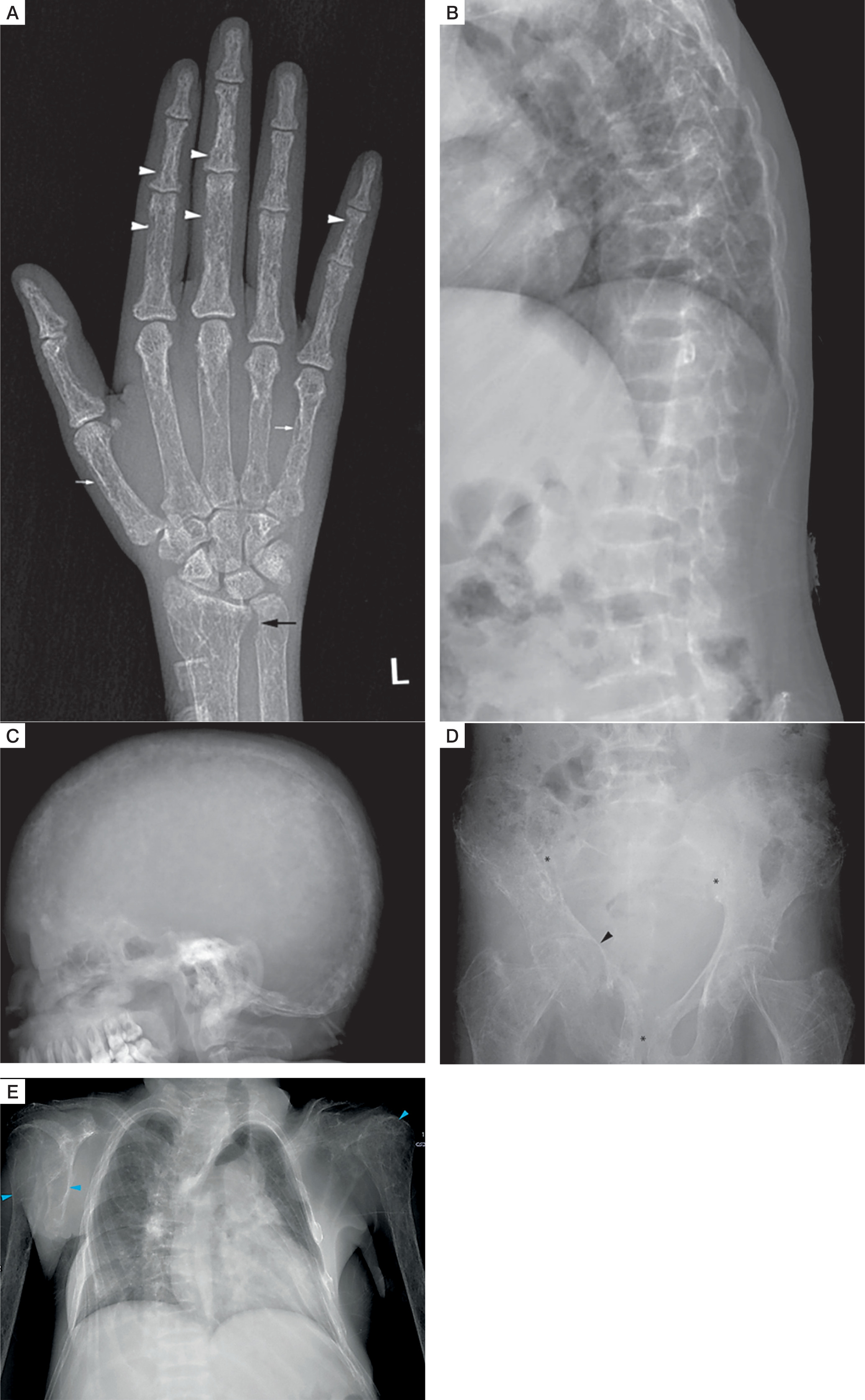
Figure 3
Non contrast computed tomography (NCCT) of kidney, ureter, and urinary bladder (KUB). Coronal (A), and axial (B) NCCT KUB images shows right ureter calculus (white arrow) causing right hydroureteronephrosis (white asterisk), and bilateral renal calculi (white arrowheads)
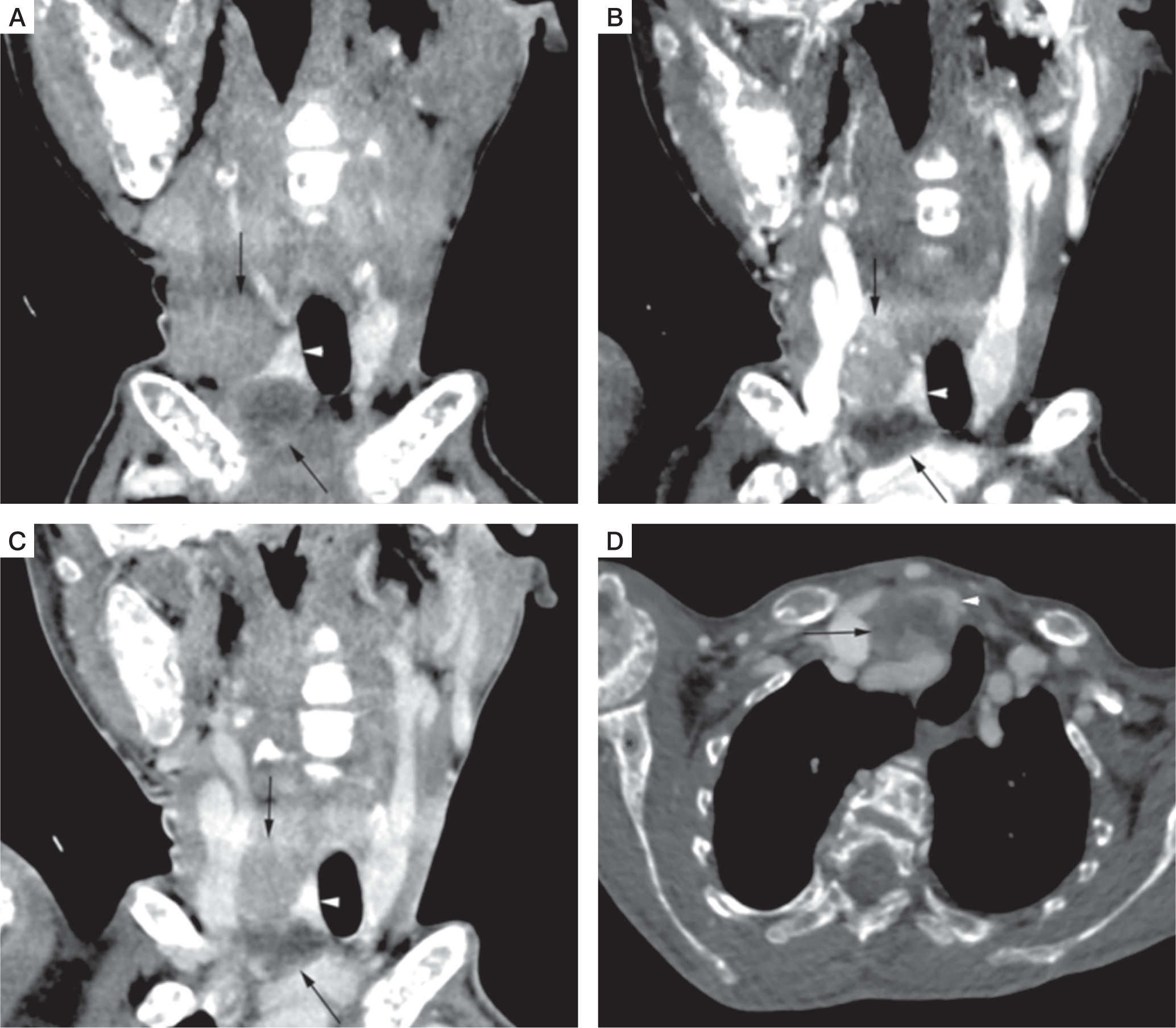
For localization of parathyroid adenoma, the patient was subjected to a 4-dimensionsal computed tomography (4D CT) scan of the neck that revealed a 3-cm right superior parathyroid adenoma and a 2.6-cm cystic right inferior parathyroid adenoma (Fig. 4A–D). There was no invasion into the surrounding structures. She also underwent a Technetium 99 SESTAMIBI scan that confirmed the location of the adenomas in the same region without any ectopic lesion.
Figure 4
4-dimensionsal computed tomography (4D CT) scan of the neck. Coronal non contrast (A), arterial phase (B), and venous phase (C) CT images showed right superior and cystic right inferior parathyroid adenoma (black arrows), deep to right lobe of thyroid gland (white arrowhead). Right superior parathyroid adenoma, and peripheral enhancing regions of cystic right inferior parathyroid adenoma appear hypodense to thyroid gland on non-contrast CT images, with enhancement in arterial phase and relative washout in venous phase. Axial venous phase CT image (D) shows cystic right inferior parathyroid adenoma (black arrow) deep to right lobe of thyroid gland (white arrowhead). Brown tumors also noted in right proximal humerus, and right scapula (black asterisks)
In the pre-operative period, hypercalcaemia was managed with intravenous saline diuresis (0.9% normal saline at 100 ml/hour followed by injectable furosemide 40 mg twice daily). Despite this, serum total calcium remained elevated (17 mg/dl). She was then treated with intravenous zoledronic acid 4 mg single dose, along with subcutaneous calcitonin 200 IU 6-hourly for 5 days. Subsequently, her serum calcium dropped to 13.8 mg/dl. Vitamin D was administered as a single dose of cholecalciferol 60,000 IU prior to surgery. This was later continued in the post-operative period with a dose of 60,000 IU once a week for 8 weeks followed by once per month.
The patient then underwent surgery. Due to poor respiratory reserve, en bloc resection of the right parathyroid with right-sided hemithyroidectomy was done under local anaesthesia. The immediate post-operative iPTH level was 15.1 pg/ml.
After surgery, the patient developed severe hungry bone syndrome. She was symptomatic with hypocalcemic symptoms like perioral numbness and carpopedal spasm. There was, however, no life-threatening complication of hypocalcaemia like arrhythmia or seizure. Her post-operative serum calcium dropped to a nadir value of 4.1 mg/dl while serum ALP raised to more than 10,000 IU/l. She required multiple infusions of calcium gluconate to recover from hungry bone syndrome. Throughout this period, she remained on our ward for 3 months. She was discharged on 6 g of elemental calcium and 2 mg of calcitriol after 3 months of surgery.
Table I provides a summary of the pre-operative, immediate post-operative, and 3-month post-operative biochemical parameters.
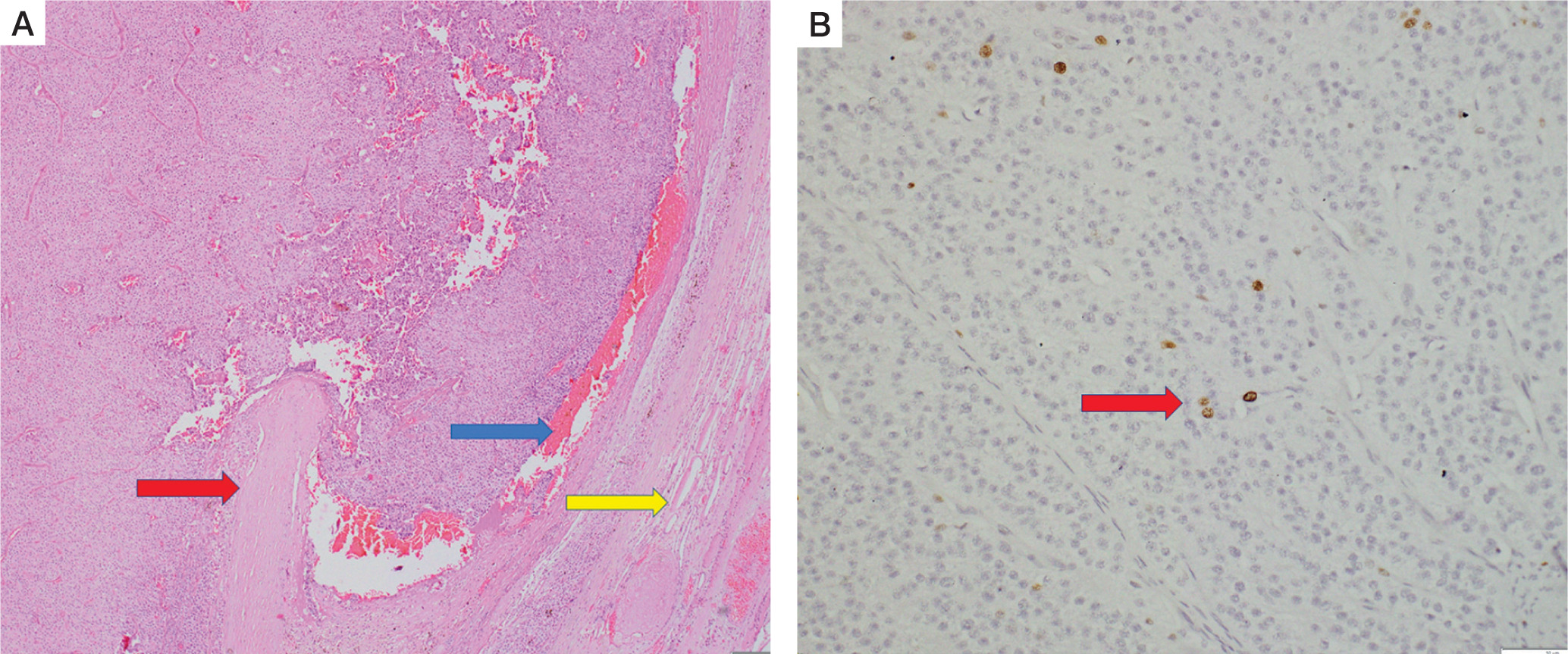
On histopathology, the specimen labelled as right superior parathyroid adenoma weighed 6 g. It was compatible with atypical parathyroid adenoma (Fig. 5A). Although the capsule of the tumour was adherent to the surrounding thyroid, there was no gross invasion into the thyroid. There was no conclusive evidence of vascular invasion. However, it did display some worrisome features like focal nuclear atypia, focal intratumoural fibrous bands, mitosis (1–2/10 high-power fields) and macronucleoli. There was no necrosis. MIB-labelling index was 2% in the areas of the highest activity (Fig. 5B).
Table I
Summary of the biochemical parameters in the pre-operative and immediate post-operative period of the patient
Figure 5
Histopathology of right superior parathyroid adenoma. Hematoxylin and eosin stain (A) shows features of atypical parathyroid adenoma. The capsule is adherent to the thyroid parenchyma (blue arrow), but gross thyroid invasion is not seen.Compressed thyroid parenchyma is noted at the periphery (yellow arrow). (B) of the tumor specimen showing positive MIB labeling index (red arrow)
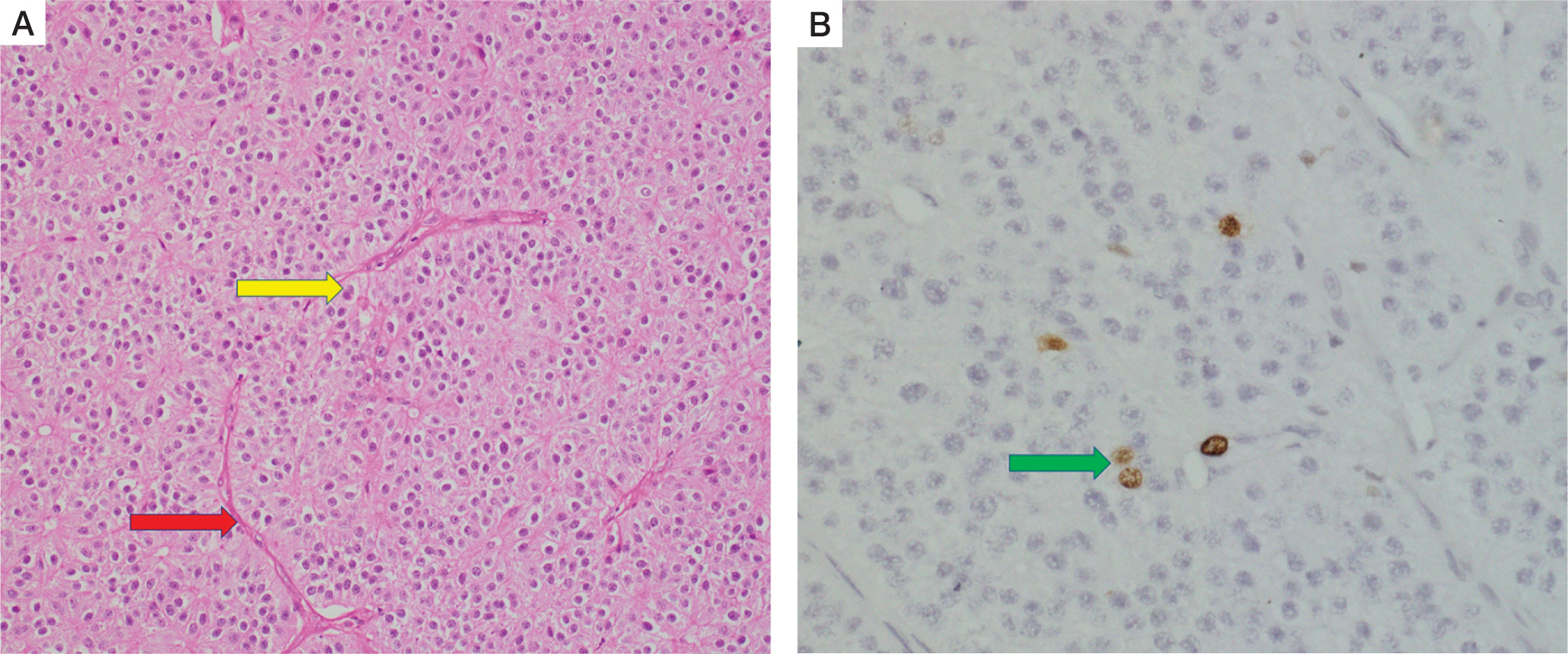
The specimen labelled as inferior parathyroid adenoma weighed 7.4 g. It was again compatible with atypical parathyroid adenoma with trabecular growth, intratumoural thick septa, cystic change, and macronucleoli (Fig. 6A). Focal oncocytic metaplasia was noted. MIB-labelling index was less than 1% in the areas of the highest proliferative activity (Fig. 6B). Normal compressed parathyroid tissue was noted at the periphery.
Figure 6
Histopathology of right inferior parathyroid adenoma. Hematoxylin and eosin stain (A) again shows features of atypical parathyroid adenoma, with macronucleoli (yellow arrow) and intervening blood vessel (red arrow) within the tumor. Immunohistochemistry (B) showing positive MIB labeling index in the tumor (green arrow)
Discussion
PHPT in children and adolescents often differs from that in adults. In children, PHPT may sometimes masquerade as rickets [4] and cause a diagnostic challenge to the treating physician. Our patient had frontal bossing and thoracic kyphoscoliosis, but other rachitic stigmata like pectus carinatum, Harrison’s sulcus, genu varum, genu valgum, or windswept deformity of lower limbs were absent. She had features of hypophosphataemic osteomalacia like proximal muscle weakness, pseudofractures, osteopaenia, and demineralization of all bones.
Our patient had marked growth retardation, weight loss, and arrested puberty. These may be attributed to the chronic systemic illness with extensive musculoskeletal, renal, and gastrointestinal involvement leading to poor nutrition and loss of appetite. Systemic illness is likely to alter the growth hormone (GH)-insulin like growth (IGF)1 axis likely to produce a GH resistant state [5]. Malnutrition can also be attributed to concomitant vitamin D deficiency, which is again more common in developing countries [6]. In children, concomitant vitamin D deficiency may mask the hypercalcaemia [4], leading to a delayed diagnosis and more severe systemic involvement. Significant short stature in paediatric PHPT has also been reported in previous case series and case reports from India [7, 8].
In children and adolescents, there is a higher possibility of multiglandular and syndromic diseases associated with PHPT. We considered the possibility of Multiple Endocrine Neoplasia type 1 (MEN1) (pituitary adenoma, PHPT, pancreatic neuroendocrine tumours), Multiple Endocrine Neoplasia Type 2A (MEN2A) (medullary thyroid carcinoma, PHPT, pheochromocytoma) Multiple Endocrine Neoplasia Type 4 (MEN 4) (pituitary adenoma, PHPT, gonadal tumours), and Hyperparathyroid Jaw Tumour (parathyroid carcinoma, ossifying fibroma of jaw). However, she did not have any symptom suggestive of syndromic involvement like galactorrhoea, headache, visual deficit, neuroglycopenic symptoms, thyroid nodule or goitre, adrenergic spells, hypertension, or jaw tumour. Due to resource constraints, genetic analysis for MEN1, RET, CDKN1B, and HRPT2 gene mutations could not be done. In the absence of genetic tests, biochemical markers for neuroendocrine tumours (NET) play a role in the diagnosis of MEN1 and MEN2A. These tumour markers include neuron-specific enolase (NSE), chromogranin A (CgA), for diagnosis of pancreatic NET in MEN1 [9], and serum calcitonin for diagnosis of medullary thyroid carcinoma in MEN2A [10]. Although these tests were not performed in our patient in the current setting, we intend to evaluate these on follow-up.
Although parathyroid carcinoma is rare in the paediatric age group, we did consider the possibility of the same based on the following grounds.
Clinical – The patient had severe bone involvement in the form of osteitis fibrosa cystica, subperiosteal resorption of digits, ‘pepper pot appearance of skull’, pathological fractures, osteopaenia, and diffuse bone pain. She had renal involvement in the form of right ureteric calculus and bilateral renal calculi. She also had an episode of acute pancreatitis. In addition, there was significant loss of weight and growth retardation along with arrested puberty. Severe bone and renal involvement led to a suspicion of parathyroid carcinoma, as reported previously [11, 12].
Biochemical – Although there is no clear threshold of biochemical parameters for diagnosis of parathyroid carcinoma, severely elevated serum calcium and serum iPTH again suggest the possibility of parathyroid carcinoma. Our patient had a baseline serum calcium level of 18.5 mg/dl and serum iPTH of more than 5000 pg/ml, which was more than 77 times the upper limit of normal (ULN) (15–65 pg/ml), and serum ALP of 2015 IU/l – again 8 times the upper limit of normal (80–240 IU/l). Serum calcium more than 14 mg/dl (or 3 mmol/l) indicates parathyroid carcinoma [11]. Similarly, iPTH more than 5–10 times the ULN and serum ALP > 300 IU/l arouse suspicion of parathyroid carcinoma [12, 13].
Radiological – On ultrasonography, parathyroid lesions more than 3 cm in size are suspicious of parathyroid carcinoma [14]. Our patient underwent a 4DCT scan that showed 2 parathyroid lesions: right superior parathyroid adenoma of 3 cm and right inferior parathyroid adenoma of 2.6 cm. Although there was no invasion into the surrounding structures, the larger size on 4DCT still favoured a possibility of parathyroid carcinoma. Schulte and Talat [12] proposed the ‘< 3 + < 3’ (serum calcium < 3 mmol/l or 14 mg/dl and parathyroid lesion size < 3 cm on USG ) rule for exclusion of parathyroid carcinoma, with a positive predictive value of 99.8% in real-life populations.
Considering a high-risk case, the patient underwent en bloc resection of right parathyroid lesions along with right hemithyroidectomy. Histopathology revealed atypical parathyroid adenomas with a few high-risk features.
The most common cause of primary hyperparathyroidism is benign parathyroid adenoma (80%), followed by parathyroid hyperplasia (15%), ectopic parathyroid adenoma (4%), and multiple parathyroid adenomas (1–2%) [15]. Parathyroid carcinomas constitute 1% while atypical parathyroid adenoma constitutes less than 1% of PHPT [14]. Atypical parathyroid adenoma falls in the intermediate spectrum between adenoma and carcinoma [15]. It has unknown malignant potential, but does not fulfil the absolute histopathological criteria for malignancy. The latter include any one of the following histopathological features: invasion of surrounding soft tissues; invasion of surrounding vital structures like thyroid, oesophagus, pharynx, larynx, trachea, recurrent laryngeal nerve, or carotid artery; vascular invasion; perineural invasion or histologically documented regional or distant metastasis [17]. In the absence of absolute criteria, the presence of the following features poses a risk of malignancy: capsular invasion (without extension into the surrounding soft tissues); readily identifiable mitotic figures (> 5/10 high-power fields); broad intratumoural fibrous bands splitting the parenchyma and separating expansile nodules; coagulative tumour necrosis; diffuse sheet-like monotonous small cells with high nucleus-to-cytoplasm ratio; diffuse cellular atypia or macronucleoli present in many tumour cells [17]. Rosai et al. defined atypical parathyroid adenoma as the presence of the following features: a) adherence to contiguous structures in the absence of clear invasion; b) banding fibrosis with or without hemosiderin deposition; c) entrapment of neoplastic cells within the fibrous capsule; d) solid, trabecular growth patterns; and e) mitotic activity [18]. In our patient, both the superior and the inferior right parathyroid adenomas exhibited high-risk features like capsular invasion, focal nuclear atypia, and intratumoural fibrous bands. However, there was no conclusive evidence of malignancy, such as invasion into the surrounding structures, vascular invasion, or regional or distant metastases.
The role of immuno-histochemical markers in atypical parathyroid adenoma is not clearly defined. There has been considerable overlap in the expression of markers like Ki67, parafibromin, galectin 3, PGP9.5, and cyclin D1 among parathyroid adenoma, atypical adenoma, and parathyroid carcinoma [16]. The molecular signature of atypical parathyroid adenomas is currently uncertain, but the germline CDC73 mutation (parafibromin) appears to be the most consistent anomaly [15]. Atypical parathyroid adenoma, in most circumstances, may be part of hyperparathyroidism jaw tumour (HPT) syndrome with CDC73 or parafibromin mutation [16]. Unfortunately, due to resource constraints, parafibromin mutational analysis could not be performed in the tumour specimens of our patient.
Clinical outcome data of atypical parathyroid adenoma have been limited and variable. There is a risk of recurrence or malignancy in the long term. In long-term studies, the overall recurrence of atypical parathyroid adenoma has been found to be 3%, which is less than that in familial forms (40%), but higher than that in sporadic adenomas (2%) [16]. The average time interval between surgery and the first recurrence is found to range from 12 months to 17 years [16].
Differentiating between atypical parathyroid adenoma and carcinoma is difficult. Long-term follow-up is required to predict the exact nature of the atypical adenoma. Annual biochemical testing and ultrasound of the neck every year after surgery are recommended to look for recurrence [19].
Conclusions
We have described a 16-year-old adolescent girl who presented with severe manifestations of primary hyperparathyroidism. Biochemically, she had severe PTH-dependent hypercalcaemia that needed management with saline diuresis, bisphosphonate, and calcitonin. Post-operatively, she developed severe hungry bone syndrome that required multiple calcium infusions over 3 months. Such severe manifestations are expected in parathyroid carcinoma. However, in our patient, final histopathology revealed an atypical parathyroid adenoma with low proliferative index, as well as diffuse sheets of small cells with increased nuclear-to-cytoplasm ratio. Our case report conveys the message that severe manifestations may be encountered even in atypical parathyroid adenomas. Long-term follow-up is required to look for any evidence of recurrence or malignancy.