

 ENGLISH
ENGLISH







Introduction
Wolcott-Rallison syndrome (WRS) (OMIM 226980) is a rare autosomal recessive disease characterized by early-onset diabetes mellitus, epiphyseal dysplasia, hepatic dysfunction, and growth retardation, and results from pathogenic variants in the Eukaryotic Translation Initiation Factor 2-Alpha Kinase 3 (EIF2AK3) gene [1]. Other clinical manifestations include hypothyroidism, exocrine pancreatic insufficiency, renal insufficiency, intellectual deficits, neutropenia, and recurrent infections [2]. Neuro-psychological development is commonly affected, although its severity is variable [1]. The neurological manifestations range from almost normal development to severe neuro-motor deficits, intractable epilepsy, intellectual deterioration, and microcephaly [1]. There are rare reports of malformations of cortical development (MCD), such as simplified gyral pattern (SGP), in children with WRS [2]. However, the lissencephaly-pachygyria pattern has not been reported as part of the clinical phenotype of WRS.
Most cases of WRS present with non-autoimmune and insulin-dependent diabetes during the first six months of age. In populations with high consanguinity rates, WRS is the most common cause of genetic neonatal diabetes mellitus (NDM) [3]. India being a multi-ethnic country, the rates of consanguinity vary widely across different regions within the country. Consequently, the occurrence of WRS varies in different geographical areas. In Southern India, WRS is considered the most common cause of NDM, while its occurrence in Northern India is rare [4, 5]. In this communication, we describe an infant from Northwest India diagnosed as WRS with additional lissencephaly-pachygyria spectrum features.
Case presentation
This 3-month-old boy was the first-born baby to non-consanguineous parents and weighed 2.6 kg at birth. At two months of age, he developed right focal seizures with varying frequency of 2–15 episodes per day, for which he was treated elsewhere with phenytoin. He was subsequently referred to our hospital after initial investigations showed hyperglycemia (random blood glucose 527 mg/dl) and lissencephaly. There was no previous history of a similar disorder in the family. The determination of lack of consanguinity between parents was made on the basis of a pedigree history.
On examination, he weighed 5.2 kg (–1.04 SDS, standard WHO growth chart) and had a small head (head circumference 38.5 cm, –2.18 SDS, standard WHO growth chart). Neurological examination revealed mild limb hypotonia and normal tendon reflexes. The rest of his systemic examination was unremarkable.
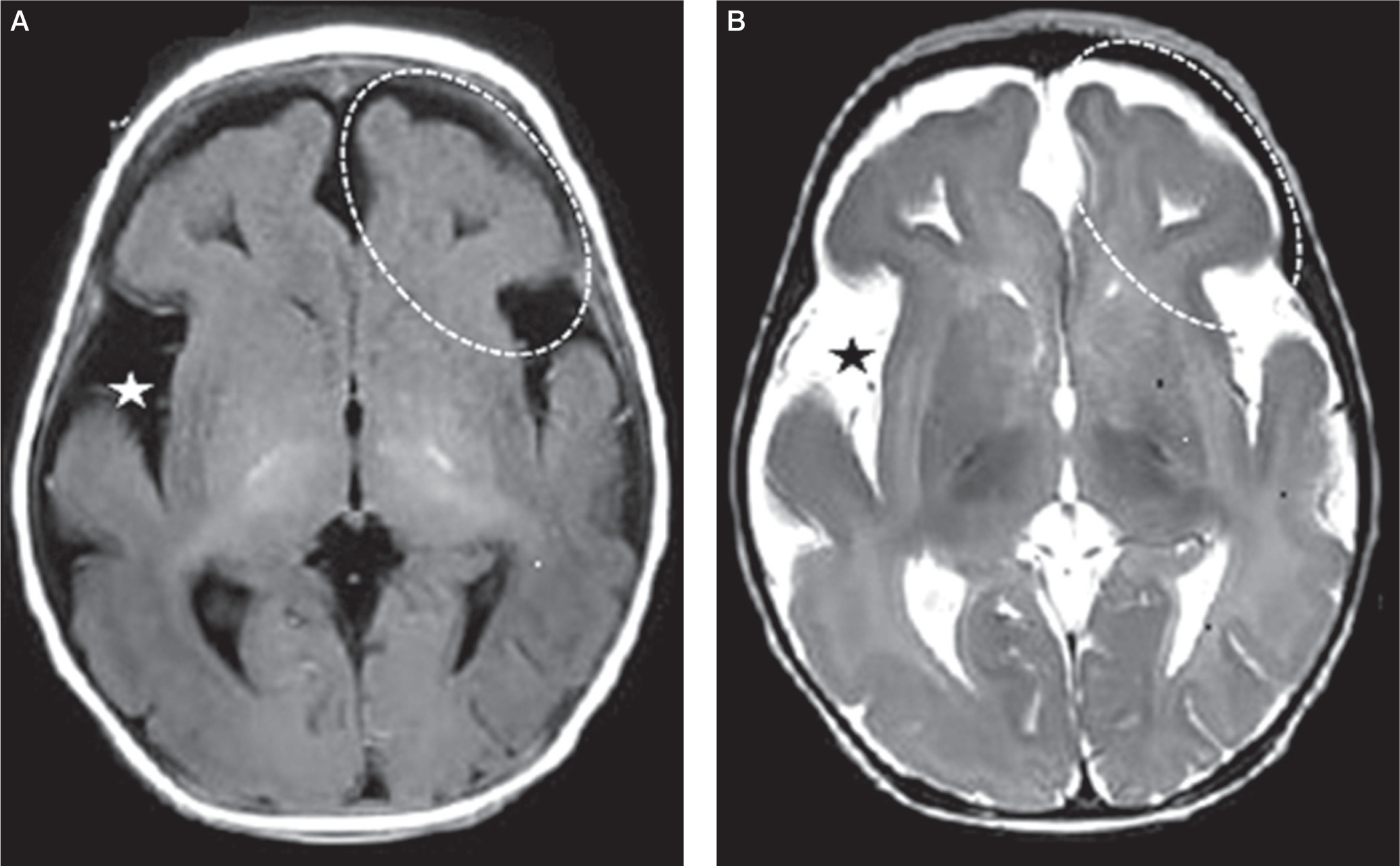
Investigations revealed hemoglobin of 10.6 g/dl, total leukocyte count 13.9 × 109/l (neutrophils 20%, lymphocytes 72%) and platelet count 358 × 109/l. The routine biochemistry, including liver and kidney function tests, were within the normal range. Glycosylated hemoglobin (HbA1c) was 13.1%, and fasting and stimulated C-peptide concentrations were 0.105 ng/ml (normal range, 0.78–1.89 ng/ml) and 0.644 ng/ml (normal range, 5–12 ng/ml), respectively. Plasma adrenocorticotropin hormone and AM cortisol concentrations were 65.35 pg/ml (normal range, 7.2–63 pg/ml) and 292.9 nmol/l (normal > 150 nmol/l), respectively. Serum triiodothyronine (T3), thyroxine (T4) and thyroid-stimulating hormone (TSH) concentrations were 0.89 ng/ml (normal range 0.2–2.0 ng/ml), 7.11 µg/dl (normal range, 4.8–12.7 µg/dl) and 3.84 µIU/ml (normal range, 0.27–4.2 µIU/ml) respectively. Pancreatic autoantibody profile showed positive islet antigen 2 (IA2) but negative anti-insulin (IAA) and glutamic acid decarboxylase 65 (GAD 65) antibodies. Electroencephalogram (EEG) showed bilateral posterior pseudo-periodic bursts of intermittent epileptiform discharges with anterior background suppression suggestive of nonspecific diffuse cortical dysfunction and underlying cortical irritative foci in left temporo-occipital region. Magnetic resonance imaging (MRI) of the brain showed anterior predominant thick cortex with SGP suggestive of lissencephaly-pachygyria complex, associated with mild cerebral atrophy and widened Sylvian fissures (Fig. 1 A, B). A monogenic NDM was considered as a provisional diagnosis.
Figure 1
Axial T1-weighted (A) and T2-weighted (B) magnetic resonance images showing anterior predominant thick cortex with simplified gyriform pattern suggestive of lissencephaly-pachygyria complex (dashes oval markers). Mild cerebral atrophy is also evident by widened Sylvian fissures (star markers)
Genomic DNA was extracted from leukocytes in the peripheral blood of the affected child and his parents. Analysis of the coding regions and conserved splice sites of 32 NDM-related genes was performed by targeted next-generation sequencing (NGS) (Agilent custom capture v5.3/Illumina NextSeq500), which can also detect partial or whole gene deletions and duplications. The NGS revealed a homozygous deletion of exon 1 in the EIF2AK3 gene at location (GRCh37, hg19) chr2:(?_88926479) (88926838_?) with HGVS description of NM_004836.5:c.(?_46)_(308+6_309 50) del, p.?. Biallelic loss-of-function variants in the EIF2AK3 gene are a known cause of WRS [6]. Confirmation testing was done with dosage analysis of exons 1 and 2 of the EIF2AK3 gene by Droplet Digital PCR using EvaGreen. Carrier testing revealed a heterozygous state for the pathogenic EIF2AK3 deletion variant in both parents. Appropriate informed consent was obtained from the parents for the laboratory studies and genetic tests. The departmental review board approved the study for publication.
The baby was initiated on a basal-bolus insulin regimen. His total insulin dose is 0.5 units/kg/day. At his latest evaluation at six months of age, he weighed 7.0 kg (–1.23 SDS), and his HbA1c was 8.8%.
Discussion
Children with WRS often present with NDM, which is an essential criterion for diagnosis [1]. Other clinical features such as skeletal dysplasia, growth retardation, hepatic, kidney, and thyroid dysfunction, neutropenia, and neurological manifestations appear as the age advances [1]. We expect that a periodic screening by radiological imaging, biochemical, hormonal, and hematological tests will reveal additional features in our patient well in time to offer appropriate management. The highlight of our case is the presence of unusual neurological manifestations in the form of the lissencephaly-pachygyria spectrum on brain imaging. Neuro-psychological manifestations are the second most common symptoms in WRS seen in almost two-thirds of patients [7]. Yet, the MCD, such as gyral pattern abnormalities, are noted rarely. One of the two previous cases, who showed MCD, was reported to have SGP on neuroimaging, while the other had pachygyria and cerebral atrophy at 21 months of age [2, 8]. Our patient is probably the youngest to show a pattern of lissencephaly-pachygyria. Interestingly, he has not shown signs of developmental regression yet. It has been observed previously that developmental regression may occur as late as 30 months of age and is particularly common after hepatic failure episodes begin to appear [7, 8]. The mechanism of neurological abnormalities is the same as the other organ dysfunctions in WRS. In normal circumstances, upon endoplasmic reticulum (ER) stress, EIF2AK3 detects the accumulation of misfolded proteins in the ER, phosphorylates EIF2α, and downregulates the rate of protein synthesis [1]. EIF2α phosphorylation results in the expression of the CHOP, a transcription factor that performs multiple functions during ER stress. The homozygous loss-of-function deletion variant in the EIF2AK3 gene makes it non-functional, resulting in the accumulation of a large amount of misfolded proteins in affected cells and tissues that leads to increased apoptosis. The increased apoptosis causes premature cell death, responsible for the organ dysfunction in WRS [1, 7].
There are some unusual features of diabetes in our patient. Typical diabetes in WRS is non-autoimmune [1, 3, 4]. The IA2 autoantibody positivity at three months of age in our patient indicates pancreatic autoimmunity, even though the main pancreatic autoantibodies commonly observed in our patient population of autoimmune diabetes were not detected [9, 10]. The frequency of pancreatic autoantibodies in the general pediatric population is extremely low, and even a single autoantibody positivity is linked with the future development of autoimmune diabetes [11]. Other characteristic features of diabetes in WRS, such as onset with DKA, poor glycemic control, and frequent occurrence of severe hypoglycemia, were not observed in our patient over the short observation period [4, 12]. The glycemic control may, however, show worsening over the long-term, similar to the experience in large patient cohorts [12].
Although WRS is a common cause of NDM in consanguineous families, its occurrence is extremely rare in NDM children born to non-consanguineous parents. None of the 11 children with genetically confirmed NDM previously reported from Northwest India had WRS [5]. There is, however, a recent report of a novel disease-causing variant of the EIF2AK3 gene in another child from our region [13]. At our center, the index patient is the second case of WRS out of 34 patients with NDM registered over the last 15 years, 12 of whom have genetically confirmed monogenic diabetes [14, 15]. This is in sharp contrast to reports from Southern India, where WRS is a common diagnosis in children with NDM, probably due to higher consanguinity rates in their population [4, 6].
In conclusion, we report a biallelic homozygous deletion of exon 1 in the EIF2AK3 gene in an infant with features of WRS that included NDM and an unusual neurological manifestation of lissencephaly-pachygyria spectrum. Screening for pathogenic genetic variants in the EIF2AK3 gene in all patients with NDM allows early diagnosis of WRS, which is vital for providing genetic counseling, optimizing management, and predicting the development of additional manifestations with this syndrome.