

 ENGLISH
ENGLISH







Introduction
Adrenocortical carcinoma (ACC) is an aggressive and rare tumour, representing 0.2% of all childhood cancers [1]. Adrenalectomy remains the first-line therapy and offers the best outcome. In contrast, the adjuvant treatment is less standardized in children [2]. The key adjuvant drug is mitotane with different chemotherapy regimens, such as etoposide, adriamycin, and cisplatin. Mitotane is an adrenotoxic drug that blocks the synthesis of hormonal corticosteroids by inhibiting multiple enzymatic steps of steroidogenesis, and it has a direct cytotoxic effect on the adrenal cortex [3]. Treatment-related side effects frequently occur. The most common adverse events are gastrointestinal and hepatic disturbances or neurological symptoms. Endocrinopathies associated with using mitotane include adrenal insufficiency (AI), precocious puberty (PP), and hypothyroidism. The symptoms can lead to temporary or permanent discontinuation of the mitotane therapy [3].
Aim of the study
The study aimed to describe patients diagnosed with PP and other endocrinological complications during mitotane treatment due to adrenocortical carcinoma, and present a literature review on the possible adverse effects of this therapy among children.
Material and methods
Patients
We report 4 patients diagnosed with ACC at The Children’s Memorial Health Institute in Warsaw (Poland). We enrolled the patients with endocrinological complications like incomplete precocious puberty during adjuvant mitotane treatment of ACC.
Clinical evaluation
The data of each patient were retrospectively analysed based on medical records: medical history, physical examination, neurological examination, results of hormonal tests before and after surgery, results of computed tomography (CT), ultrasound examinations, and histopathological, immunohistochemical, and genetic studies.
Luteinizing hormone (LH), follicle-stimulating hormone (FSH), oestradiol, testosterone, androstenedione, dehydroepiandrosterone sulphate (DHEAS), prolactin (PRL), cortisol, thyroid-stimulating hormone (TSH), free triiodothyronine (FT3), and free thyroxine (FT4) were measured with the chemiluminescent immunoassay method (Alinity I/Abbot). In Patient 1, LH, FSH, oestradiol, testosterone, androstenedione, and DHEAS were analysed with the radioimmunoassay method.
Until May 2021, adrenocorticotropic hormone (ACTH) was measured with the radioimmunoassay method, and then with the chemiluminescent immunoassay method on the Cobas e411/ Roche analytical system. The 24-hour UFC excretion was evaluated by the gas chromatography-mass spectrometry (GC-MS) method, with the reference range varying by age.
Surgical procedures
All patients underwent lateral adrenalectomy. In 2 cases this was carried out by laparotomy, and in 2 cases by laparoscopy. After one year of follow-up, patient 2 underwent right adrenalectomy due to the occurrence of a metastatic lesion.
Histopathological evaluation
Each of the tumours was examined in its entirety so as to reveal subsequent cross-sections of the tumour along with the surrounding tissues. All resected specimens were subjected to a uniform protocol for formalin-fixed, paraffin-embedded (FFPE) specimens. All the haematoxylin-eosin (H&E) slides of the surgical specimen were reviewed and examined by 2 experienced pathologists. The tumours were graded into malignancy based on criteria according to the 2022 WHO classification of adrenal cortical tumours.
Immunohistochemical studies were performed on all the formalin-fixed paraffin-embedded sections using an automated stainer Ventana BenchMark ULTRA (Roche Diagnostics) system with Ventana and Celle Marque primary antibodies.
Genetic research
In 3 patients, germline TP53 gene mutation was evaluated; in 2 of them, tests for detecting somatic mutation of the TP53 gene were also performed. Samples were analysed by direct Sanger sequencing.
Results
All 4 patients were female. The median age at the diagnosis was 1.5 years (minimum [min]: 0.75 years, maximum [max]: 7.5 years). The first symptoms were hyperandrogenic signs in the form of the appearance of pubic hair and, in 2 cases, clitoromegaly. Any manifestations of cortisol excess were observed.
The median time between the first symptoms and diagnosis establishment was 1.5 months (min: 0.25 months, max: 5 months).
Abdominal CT was performed in all cases and revealed an adrenal tumour with a median diameter of 50 mm (min: 49.6 mm, max: 70 mm). The patients underwent lateral adrenalectomy, and histopathological examination confirmed ACC. All patients were treated with mitotane adjuvant therapy and, in 2 cases, with other chemotherapy regimens. The median time from surgery to the initiation of mitotane therapy was 26 days. During mitotane treatment, PP was confirmed based on symptoms, and hormonal and imaging tests. The median time from the therapy initiation to the first symptoms of PP was 4 months. The median duration of mitotane treatment was 10.5 months.
Case 1
A 7.5-year-old girl was admitted to the Department of Endocrinology because of pubic hair and a tumour of the left adrenal gland (6–7 cm in diameter, with foci of necrosis, enlargement of the adrenal vein) detected by abdominal CT.
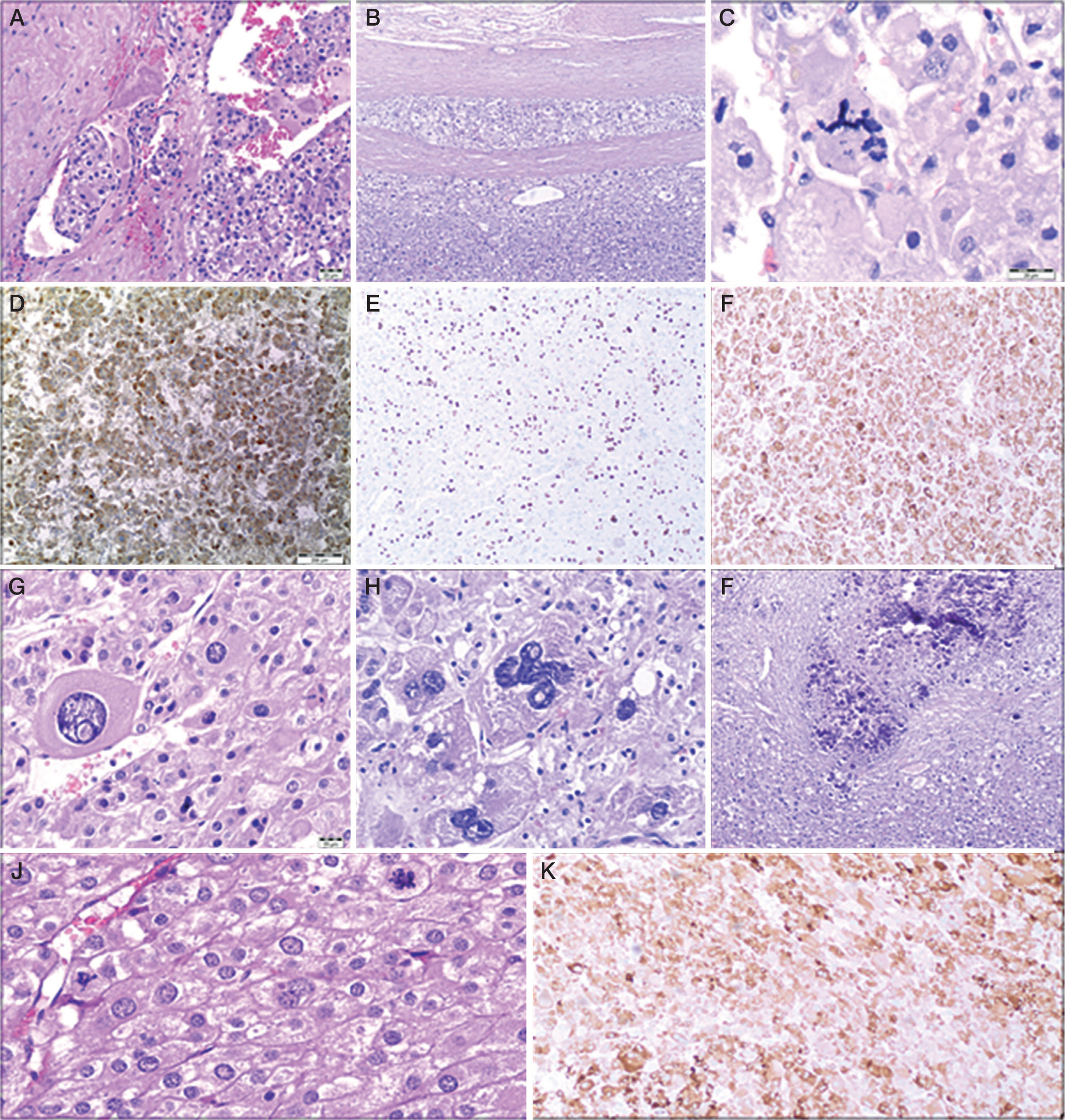
Upon admission, the patient’s height was 125.0 cm (25th–50th percentile, –0.28 SD), and her weight was 22 kg (25th percentile, –0.78 SD). Tanner’s stage 3 – pubic hair (P) and 1 – breast (B) were observed (pubic hair appeared 5 months prior). The laboratory tests revealed increased DHEA-S, androstenedione, testosterone concentrations, and normal serum cortisol value (Table I). A steroid profile in urine showed an excess of steroid metabolites. The patient underwent a left-sided adrenalectomy carried out by laparotomy. Histopathological examination confirmed adrenocortical carcinoma. Unfavourable histological features were present, including focally significant cell polymorphism, vascular (Figure 2A) and capsular invasion (Figure 2B), with few clear cells.
Table I
Results of laboratory tests at diagnosis (I) and post-surgery (II)
The tumour was classified as stage III according to the European Network for the Study of Adrenal Tumours (ENSAT) staging classification (T3, Nx, M0). One week after surgery, the patient was administered adjuvant therapy with mitotane initiated at a dose of 500 mg/day (0.58 g/m2/day). It was gradually increased to 6 g/day with serum level monitoring (serum concentrations ranged from 3.89 to 50 µg/ml, and most of the treatment time was maintained within a target range). A supraphysiological dose of hydrocortisone (17.4 mg/m2/day) was also prescribed. Adrenal androgen concentrations controlled postoperatively were within normal limits (Table I).
Three months after surgery, the patient was readmitted to the Endocrinology Department due to early breast development. Tanner’s stage P3, B2, and nipple hyperpigmentation were assessed. Pelvic ultrasonography showed 2 cm ovaries, with 4 mm in diameter follicles in both and an endometrium of 4 mm. The patient reported general weakness, vomiting, and drowsiness. Additionally, headaches, dysarthria, apathy, lethargy, low mood, vision impairment, and muscular hypotonia were observed. Adrenal androgens concentrations were within the normal range, and the gonadotropin-releasing hormone (GnRH) stimulation test confirmed GnRH-independent PP. Her oestradiol level was low. The mitotane concentration was 36.86 µg/ml, and the dosage was decreased.
Vaginal bleeding appeared after 9 months of treatment, with the blood concentration of mitotane 46.35 µg/ml (dosage 4 g/day). Repeated GnRH stimulation testing revealed that she had developed a secondary central precocious puberty (CPP). Oestradiol concentration was in the normal range (7.7 pg/ml), and her bone age was slightly advanced – 9 years (chronological age: 8.3 years). The dose of mitotane was reduced to 1.5 g/day, maintaining the target range of drug concentration. Brain magnetic resonance imaging (MRI) showed no pituitary abnormalities. To suppress the progression of puberty, a GnRH analogue (triptorelin) treatment was introduced, which has been continued for 2.5 years. The patient had menarche at the age of 11.4 years and reached normal final height in accordance with mid-parental height target. The patient was treated with mitotane for 5 years. After months of mitotane cessation, the girl completely recovered from AI, and hydrocortisone (HC) treatment was withdrawn. Neurological symptoms regressed about 6 months after discontinuation of mitotane. After 30 years of follow-up, the patient was recurrence-free.
Case 2
An 18-month-old girl was admitted to the Endocrinology Department due to an enlargement of the clitoris, development of pubic hair, and rapid weight gain (1 kg per month). The patient’s height was 80.5 cm (25th–50th percentile, –0.27 SD), and weight was 13.6 kg (90th–97th percentile, +1.95 SD). Tanner’s stage P3 and B1 were observed (pubic hair appeared one month prior). Abdominal CT revealed the presence of a 50 × 40 × 30 mm tumour in front of the left kidney with the left adrenal gland partially visible; no enlarged lymph nodes were visible (Figure 1.1A–D). Laboratory tests showed a significantly elevated concentration of DHEA-S, androstenedione, 17-hydroxyprogesterone (17-OHP), and testosterone (Table I). Circadian cortisol rhythm, oestradiol, LH, and FSH were within normal range. A steroid profile in urine showed an excess of steroid metabolites. Left-sided laparoscopic adrenalectomy was performed, and the pathological diagnosis of high-grade ACC was confirmed. Histopathological features like cytologic atypia, the presence of foci of necrosis, angioinvasion, and infiltration of the tumour capsule were observed. Mitotic activity was approximately 30 mitoses/50 HPF, including single atypical mitoses (Figure 2c). Immunohistochemical examination revealed the following: AE1/3(–), synaptophysin (+) (Figure 2D), vimentin (+), CD34(–), epithelial membrane antigen (EMA) (–), chromogranin (+) focally, Ki-67 up to 40% positive tumour cells (Figure 2E), and Melan A (+) (Figure 2F). The tumour capsule raptured during the resection, but inside the retrieval bag. The tumour could not be classified according to ENSAT staging classification (T1?, Nx, M0). Adjuvant chemotherapy with EDP (etoposide, doxorubicin, and cisplatin) was introduced, and after the treatment a CT of the abdomen and pelvis revealed no abnormalities. Control levels of adrenal androgens, cortisol, and ACTH were normal.
Figure 1
Axial unenhanced abdomen computed tomography (CT) scans (1A, 1C, 2A, 3A) and enhanced CT scans (1B, 1D, 2B, 3B) demonstrate adrenal tumours in our cases (min, max, and mean depict the respective values for the lowest, highest, and mean Hounsfield units (HU), as derived from the software for the region of interest marked by the investigator). A) Case 2, axial unenhanced CT scan demonstrates tumour act in the left adrenal gland, dimensions: 50 × 40 × 30 mm, mean 42 HU. B) Case 2, enhanced CT scan demonstrates tumour in the left adrenal gland, dimensions: 50 × 40 × 30 mm, mean 79 HU. C) Case 2 –axial unenhanced CT scan demonstrates metastases in the right adrenal gland, dimensions: 27 × 29 × 36 mm, mean 35 HU. D) Case 2, axial enhanced CT scan demonstrates metastases in the right adrenal gland, dimensions: 27 × 29 × 36 mm, mean 81 HU. E) Case 3, axial unenhanced CT scan shows an inhomogeneous tumour in the right adrenal gland, dimensions: 53 × 43 × 59 mm, ∅ 50 mm, mean 31 HU. F) Case 3, axial enhanced CT scan shows an inhomogeneous tumour in the right adrenal gland, dimensions: 53 × 43 × 59 mm, ∅ 50 mm, mean 83 HU. G) Case 4, axial unenhanced CT scan shows an inhomogeneous tumour, with peripheral calcifications in the left adrenal gland, dimensions: 49 × 39 × 45 mm, ∅ 45 mm, mean 33 HU. H) Case 4, axial enhanced CT scan shows an inhomogeneous tumour with peripheral calcifications in the left adrenal gland, dimensions: 49 × 39 × 45 mm, ∅ 45 mm, mean 94 HU
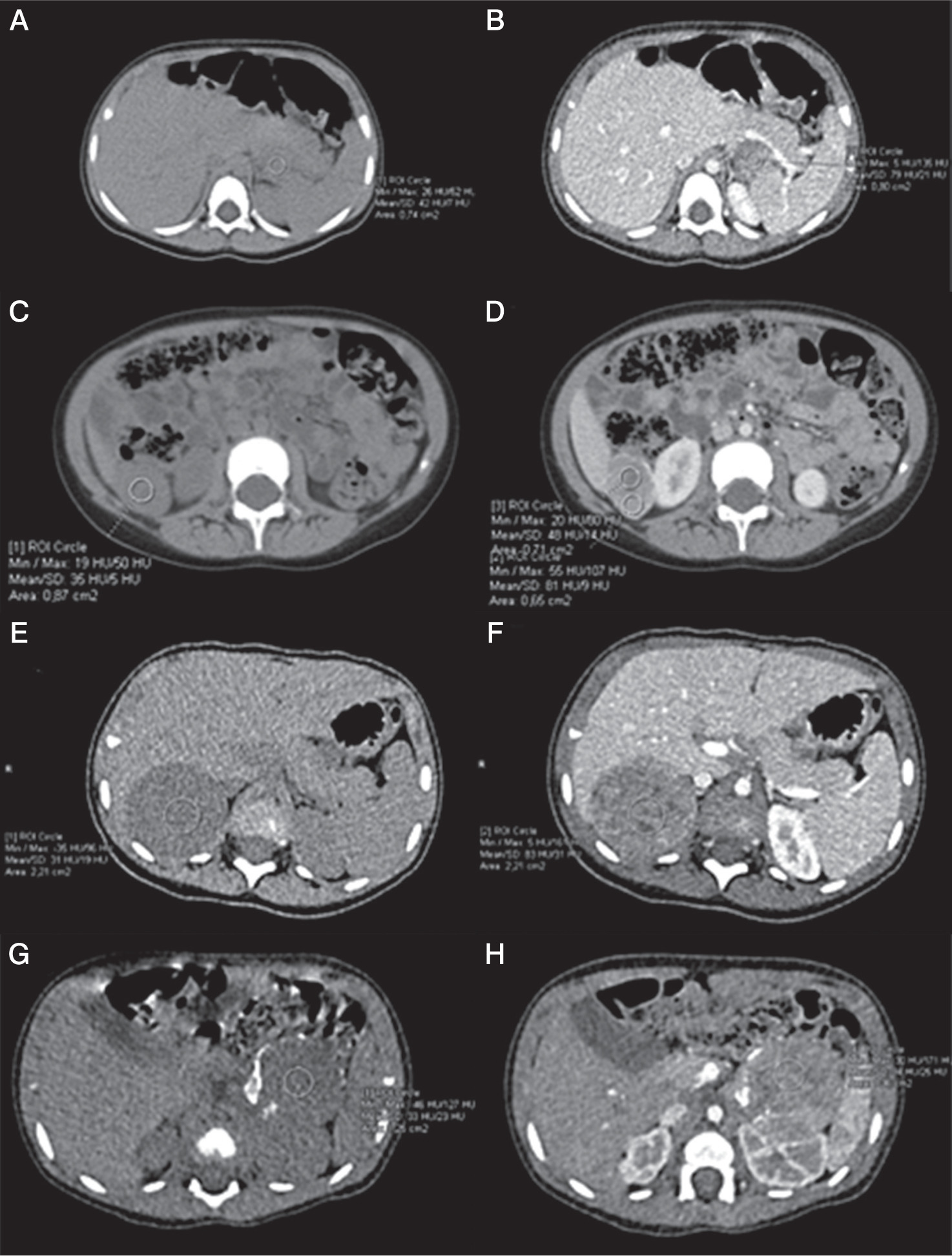
Figure 2
Histopathological examination. A) A group of cancer cells is visible in the lumen of one of the vessels. Haematoxylin and eosin (H&E) stain. B) Tumour cells infiltrating the tumour capsule made of connective tissue. H&E stain. C) Atypical mitotic figure. H&E stain. D) Immunohistochemistry examination revealed synaptophysin expression in tumour cells. E) High mitotic activity in tumour cells. Ki-67 positive tumour cells. (Immunohistochemistry staining). F) Immunopositivity for Melan A in tumour cells. G, H) Polymorphic cells. Cells of various shapes and sizes, including bizarre cells. H&E stain. I) A focus of neoplastic necrosis with the presence of calcifications. H&E stain. J) High mitotic activity in tumour cells. Three mitotic figures in one high-power field (HPF). H&E stain. K) Immunohistochemistry examination revealed Inhibin expression in tumour cells
At the age of 2.5 years (one year after the surgery), in the control CT examination, an abnormal mass was described between the lower pole of the right kidney and the liver measuring 27 × 29 × 36 mm. The concentrations of DHEA-S, testosterone, and androstenedione were significantly elevated. The patient was qualified for the surgery. A histopathological examination confirmed metastatic ACC. A genetic examination revealed germline mutation c. 538G>A p.(Glu180Lys) in exon 5 of TP53 gene. The patient was administered adjuvant therapy (chemotherapy and mitotane). Mitotane was initiated at a dose of 1 g/day (1.78 g/m2/day). It was gradually increased to 2.5 g/day with serum level monitoring (maximum serum drug concentration: 19.7 µg/ml, most of the treatment time was maintained within a target range).
A substitutive dose of hydrocortisone (12.5–16.40 mg/m2/day) and fludrocortisone (0.025–0.075 mg/day) was administered for treatment. In addition, hypothyroidism was diagnosed (TSH 2.14 uIU/ml (N: 0.67–4.16 uIU/ml), fT4 0.69 ng/dl (N: 0.8–1.7 ng/dl), fT3 2.93 pg/ml (N: 1.45–3.5 pg/ml), and anti-thyroid antibodies were negative. The patient required treatment with levothyroxine at a dose of 1.0 µg/kg/day.
Five months after the start of mitotane therapy, the patient developed symptoms of adrenal crisis: weakness, nausea, vomiting, and 2 kg weight loss. In physical examination, dry, grey skin and excessive pigmentation of nipples were noticed. Laboratory tests revealed hyponatraemia (127 mmol/l) and hyperkalaemia (5.9 mmol/l). The dose of hydrocortisone was increased from 16 mg/m2/day to 40 mg/m2/day, and the treatment with fludrocortisone was adjusted accordingly, improving the patient’s condition.
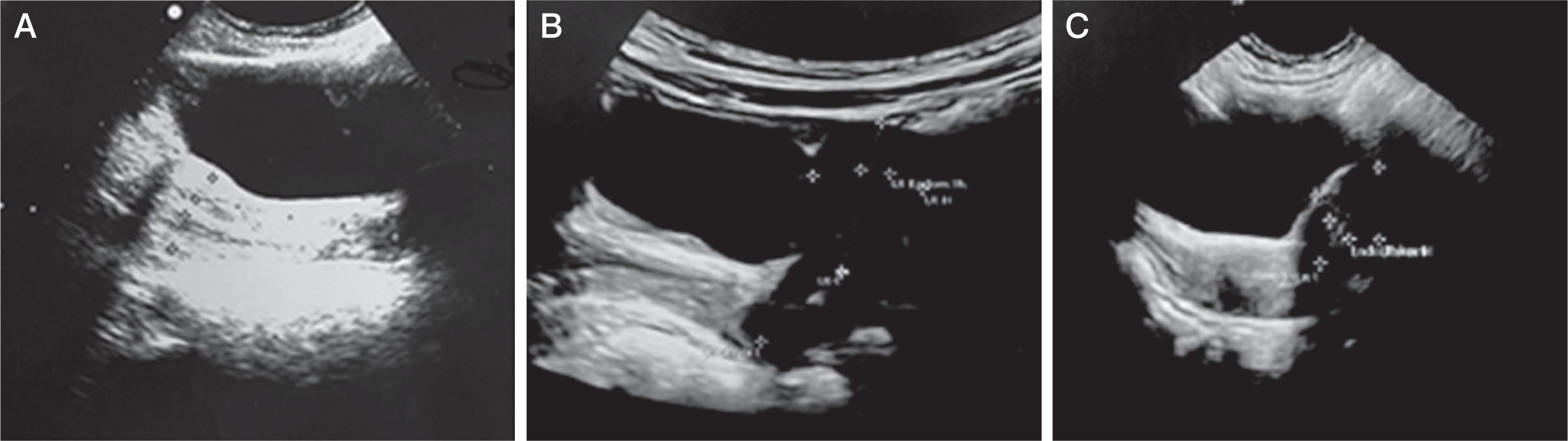
After more than 9 months of mitotane treatment, the 3.5-year-old girl was readmitted to the Endocrinology Department due to vaginal bleeding. Tanner’s stage was assessed as P1, B2. A GnRH test was performed, and the result was prepubertal. The oestradiol concentration was within the normal range. Pelvic ultrasonography showed an anteverted uterus with an anteroposterior (AP) diameter of 15 mm and a length of 26 mm. The endometrium measured 3.5 mm, and the cervical length was 23 mm. The right ovary measured 18 × 10 × 17 mm with a volume of 1.7 ml (no larger follicles were observed) (Figure 3A). The left ovary could not be visualized.
Figure 3
Pelvic ultrasound performed during mitotane treatment presents signs of precocious puberty. A) Case 2: anteverted uterus with an anteroposterior (AP) diameter of 15 mm, and a length of 26 mm. The endometrial thickness is 3.5 mm, and the cervical length 23 mm. B) Case 3: anteverted uterus with AP dimensions of 13 mm, length of 24 mm, cervix length of 21 mm, and endometrial thickness of 4 mm. C) Case 4: anteverted uterus with AP diameter 14 mm, length 20 mm, and endometrial thickness 4.8 mm
Additionally, asymmetrical development of breast tissue was noted. A recurrent episode of vaginal bleeding occurred one month after hospitalization, which lasted 3 days. Total body MRI did not reveal any signs of tumour regrowth but described an enlarged uterus with an endometrial thickness of 4 mm. Due to mitotane treatment complications and neoplastic disease remission, it was decided that mitotane would be discontinued after 12 months. After mitotane cessation, there was no progression of maturation.
During mitotane treatment, we also observed speech regression, dysarthria, stuttering, apathy, irritability, and excessive sleepiness in the patient. Symptoms resolved about 3–4 months after discontinuation of mitotane. Currently, she is experiencing symptoms of impaired concentration and memory disorders, which may also be related to her chemotherapy history. The follow-up period is 7.2 years, with no biochemical or radiological features of recurrence for 6 years.
Case 3
A 1.5-year-old girl was admitted to the Department of Endocrinology due to symptoms of hyperandrogenism (pubic hair and clitoromegaly), observed for 2 months. Upon admission, her height was 78 cm (25th percentile, –1.42 SD), and her body weight was 11.2 kg (50th percentile, +0.13 SD). Tanner’s stage was assessed as P2, B1. Enlarged labia majora with dark pigmentation, enlarged clitoris, and oestrogenisation of the vaginal mucosa was noted. DHEA-S, androstenedione, 17-OHP, and testosterone levels were significantly elevated. The diurnal cortisol rhythm was abnormal, while the ACTH level was at the lower limit of normal (Table I). The oestradiol concentration was in the normal range: 17.02 pg/ml. A steroid profile in urine showed an excess of steroid metabolites. In the examination of catecholamines and their metabolites in urine, a slightly elevated level of homovanillic acid (HVA) at 1.6–2 mg/24 h (N < 1.0) was found. Abdominal ultrasound and CT scan revealed a heterogeneous tumour in the right adrenal gland, approximately 50 mm in diameter, bulging into the liver and compressing the upper part of the right kidney (Figure 1.2A, B). A right laparoscopic adrenalectomy was performed. Histopathological examination revealed adrenal cortical carcinoma composed of pleomorphic cells (Figure 2G, H) with increased mitotic activity and atypical mitoses. Necrotic areas (Figure 2I), angioinvasion, and loss of tumour capsule continuity were present. Tumour invasion was present in the kidney capsule and perinephric fat tissue. Immunohistochemical staining showed Melan A (+), β-catenin (–), p53 (–), α-thalassemia/mental-retardation-syndrome-X-linked (ATRX) (+), and mitotic activity at approximately 50%. The tumour was classified as stage III according to ENSAT staging classification (T3, Nx, M0). Genetic testing confirmed TP53 somatic and germline mutations - heterozygous carrier status for the molecular variant: c.455C>T, p.(Pro152Leu). Serum androgen levels measured 9 days after surgery were within the normal range (Table I). 18F-fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG PET/CT) scan showed no evidence of highly metabolically active disease. After the surgery, hydrocortisone replacement therapy was introduced, gradually increasing (the dose to 18.2 mg/m2/day). Adjuvant chemotherapy with EDP, and mitotane was chosen. Mitotane administration started 1.5 months after surgery, initially at a dose of 750 mg/day (1.44 g/m2/day) and then gradually increased while monitoring the drug concentration in the blood, up to a dose of 3.5 g/day.Four months after starting mitotane treatment, the girl’s mother reported increased vaginal discharge and hyperpigmentation of nipple-areola. Within the next month, the right breast tissue developed. Additionally, the patient began experiencing tremors in the limbs, unsteady gait, and leg pain. A pelvic ultrasound revealed an anteverted uterus with dimensions of 13 mm (AP), 24 mm (length), cervix length of 21 mm, endometrial thickness of 4 mm, and right ovary measuring 14 × 9 mm and left ovary measuring 9.5 × 6 mm (Figure 3B). During this period, the maximum serum concentration of mitotane was 17 µg/ml (3.5 g/day). The dose was reduced to 3 g/day. The patient was readmitted to the Department of Endocrinology at 2 years and 5 months. Physical examination revealed breast tissue development, hyperpigmentation of the perineal area, swelling of the labia majora with signs of oestrogenisation, and white vaginal discharge. According to the Tanner scale, the patient was P1, B2. The mitotane level was 11.5 µg/ml. A GnRH test was performed, and the result indicated a prepubertal state. The oestradiol level was 14.08 pg/ml (N < 11.8–62.7). Bone age was accelerated, corresponding to 3 years and 9 months (chronological age: 2.4 years). A whole-body CT scan did not show any signs of tumour regrowth. A neurological examination confirmed weakened tendon reflexes in the lower limbs and dysmetria in the upper limbs. Considering the overall clinical status and adverse symptoms, the decision was made to discontinue mitotane after 9 months of treatment. Neurological symptoms have largely regressed about a month after discontinuation of mitotane. There are only limb tremors, only with precise movements. The follow-up period is 1.1 years, with no features of recurrence.
Case 4
A 9-month-old girl presented with the appearance of pubic hair and an enlarged clitoris for about a week. Upon admission she had a body length of 77 cm (> 97th percentile, +2.71 SD) and a body weight of 11.9 kg (90–97th percentile +3.79 SD). The Tanner stage was assessed as P2, B1. Significantly elevated levels of adrenal androgens were observed in the laboratory tests (Table I). Additionally, circadian cortisol rhythm was normal, neuron-specific enolase (NSE) 27.98 µg/l (N < 18.3). A urinary steroid profile indicated massive excretion of dehydroepiandrosterone and its metabolites and increased excretion of cortisol metabolites. Two 24-hour urine collections for catecholamines and their metabolites were also performed, revealing slightly elevated levels of noradrenaline in one collection (12.6 µg/24 h, N < 12.2), as well as elevated levels of HVA (1.6 mg/24 h, N < 1.0) and VMA (1.1 mg/24 h, N < 1.0) in both collections. Abdominal ultrasound and CT scan revealed an abnormal, heterogeneous, well-defined, calcified mass measuring 4.5 cm below the tail of the pancreas and anterior to the left kidney (Figure 1.3A, 1.3B). The patient underwent a left-sided adrenalectomy carried out by laparotomy. A histopathological examination revealed adrenocortical carcinoma with cytologic atypia, area of necrosis, focal capsular infiltration, high mitotic activity (Figure 2J), and angioinvasion. Immunohistochemical analysis showed focal NSE (+), p53 (+), weak synaptophysin reactivity, Melan A (+), inhibin (+) (Figure 2K), and focal Ki-67 staining in up to 25% of cells. The tumour was classified as stage II according to ENSAT staging classification (T2, N0, M0). Follow-up serum androgen levels were within the normal range. 18F-FDG PET/CT scan showed no evidence of highly metabolically active disease. Genetic testing confirmed the TP53 gene somatic and germline mutation c.706T>G, p.(Tyr236Asp). Three months after the surgery, treatment with mitotane was initiated at an initial dose of 500 mg (0.9 g/m2/day), gradually increasing to 3 g/day. Controlled blood levels of mitotane ranged from 3.1 to 15.1 µg/ml. HC was also administered at 10 mg/day (18 mg/m2).
At 16 months of age (4 months after starting mitotane treatment), the girl was readmitted to the Department of Endocrinology due to breast gland development and reoccurrence of pubic hair growth observed for the past 2 weeks. Regression of development was also noted; she stopped walking and spoke less. She experienced limb tremors and had an unsteady gait. Furthermore, the parents reported daytime sleepiness, night terrors, anxiety, decreased appetite, and weight loss. The puberty stage was assessed as P2 (sparse hair), B2. Labial hypertrophy, hyperpigmentation in the posterior labial commissure, and hyperpigmentation of the nipple-areolar complex were noticed. Laboratory tests revealed a decreased serum sodium level (133 mmol/l) with a potassium level of 5.36 mmol/l and an ACTH level of 671 pg/ml, and an undetectable cortisol level before administration of the next dose of HC. The HC dose was increased to 15 mg/day (27.3 mg/m2). Based on the evaluation of thyroid function, hypothyroidism was diagnosed (TSH 6.5 uIU/ml (N: 0.87–6.15 uIU/ml), fT4 0.77 ng/dl (N: 0.94–1.44 ng/dl), fT3 3.63 pg/ml (N: 3.3–5.2 pg/ml), and anti-thyroid antibodies were negative. The patient required treatment with levothyroxine at a dose of 1.13 µg/kg/day. Pelvic ultrasound revealed the following: uterine body length 20 mm, anterior-posterior diameter 14 mm, endometrial thickness 4.8 mm, right ovary volume 2.4 ml, left ovary volume 3.7 ml, and multiple follicles in the ovaries (Figure 3C). A GnRH test resulted in prepubertal findings with an oestradiol concentration of 21.30 pg/ml (N < 11.8–62.7). Bone age was assessed as 1 year and 6 months (consistent with chronological age). The controlled mitotane level was 21.4 µg/ml. Neurological examination revealed features of cerebellar ataxia. After 4 months of mitotane treatment, due to the increased severity of side effects associated with mitotane, including PP and neurological disturbances, the decision was made to discontinue the mitotane administration. Neurological symptoms have largely regressed about a month after mitotane cessation; there are only limb tremors, and only with precise movements. The follow-up period is 9 months with no features of recurrence.
Discussion
We present 4 patients with peripheral/central precocious puberty during adjuvant mitotane treatment of adrenocortical carcinoma. In addition to the symptoms of PP, the patients presented with AI, hypothyroidism, and neurological complications.
ACC is a rare but very aggressive childhood cancer with an annual worldwide incidence of 0.3–0.38 cases/million children below 15 years old, with female preponderance (0.2% of all cancers diagnosed in children) [4, 5]. About 65% of adrenocortical tumours occur at < 5 years of age [6]. Of note, in southern Brazil, the incidence of paediatric ACC is 10–15 times higher than the worldwide occurrence; this is thought to be related to a specific germline mutation at codon R337H in the TP53 gene [7, 8]. Only about 20% of paediatric adrenocortical tumours (ACTs) are classified as adrenocortical adenomas (ACA), and they are associated with an excellent prognosis. A total of 80–90% of cases are hormonally active, responsible for virilization, Cushing syndrome (CS), puberty anomalies (GnRH independent precocious puberty – iso- or hetero-sexual pattern), and hyperaldosteronism [9]. Most young patients present with virilization (pubic hair, accelerated growth, skeletal maturation, an enlarged penis or clitoris, hirsutism, and acne) due to excess androgen secretion alone or in combination with cortisol in more than 80% of patients [1].
The first symptoms in the study group were hyperandrogenic signs in the form of the appearance of pubic hair and clitoromegaly. No manifestations of cortisol excess were observed. The median age at the time of diagnosis was 1.5 years. The median time between the first symptoms and diagnosis establishment was 1.5 months (min: 0.25, max: 5 months). The median tumour diameter in CT examination was 50 mm (min: 49.6 mm, max: 70 mm) (Figure 1).
A total of 50–80% of children have genetic/epigenetic anomalies involving tumour protein TP53, IGF2 overexpression, IGF1R, ZNRF3, CTNNB1, menin, PRKAR1A, chromosome 11 anomalies (11p15), and a recently discovered a mutation in the EGFR gene. Hereditary syndromes associated with adrenocortical carcinoma include Li-Fraumeni, Beckwith-Widemann, Carney syndrome, multiple endocrine neoplasia type 1 (MEN1), familial adenomatous polyposis (FIPA), and Lynch syndrome [10].
Genetic testing is recommended before treatment. Germline mutation in the TP53 gene was found in 3 patients of our study group. Mutations were also confirmed in the fathers of 2 girls (one of them was Argentinian). In the first patient diagnosed in 1992, genetic testing was not performed.
Adrenalectomy is the first-line therapy, offering the best prognosis if complete removal is achieved. According to ESMO-EURACAN (European Society for Medical Oncology – European Reference Network for Rare Adult Solid Cancers) guidelines, complete en bloc resection of all adrenal tumours suspected to be ACC by a surgeon experienced in adrenal and oncological surgery is the mainstay of a potentially curative approach. Open surgery is the standard treatment, but in small tumours (< 6 cm) without evidence of local invasion, laparoscopic adrenalectomy is reasonable if the surgeon has sufficient experience. Additionally, a locoregional lymphadenectomy is suggested [11, 12]. The preferred surgical approach in children should be an open approach. Minimally invasive surgery is strongly discouraged when malignancy is strongly suspected. Minimally Invasive techniques in the surgical management of ACC are not recommended, even when feasible. They should only be considered in early childhood, and their use should be limited to small-volume localized tumours that are probably benign without invasion of surrounding structures and nodal involvement at preoperative imaging [13].
All patients underwent adrenalectomy – in 2 cases carried out by laparotomy with lymphadenectomy and in 2 patients by laparoscopy.
Even after complete resection, a high risk of recurrence of ACC remains. Age > 4 years, tumour size, presence of Cushing syndrome at diagnosis, incomplete resection, and advanced stage have commonly been reported as adverse prognostic factors. It has been almost universally observed that children under 4 years old have a better outcome than adolescents. A recent work published by the European Group (EXPeRT) tried to define “high-risk” tumours based on the presence of one of the following features: age at diagnosis, volume more than 200 cm3, Cushing syndrome, initial biopsy, surgical excision with microscopic residuals or spillage (R1) or macroscopic residuals (R2), regional lymph node involvement, histologic vascular invasion, and distant metastases at diagnosis [14].
However, for localized disease, surgery alone is recommended; in the International Paediatric Adrenocortical Tumours Registry (IPACTR), 12% of patients with stage I or II received adjuvant chemotherapy, including EDP and mitotane [6]. In a cohort of 111 children with ACC from the National Cancer Database (NCDB), adjuvant systemic therapy was utilized in 30% of children who underwent surgical resection [15].
Chemotherapy with vincristine, ifosfamide, carboplatin, etoposide, and doxorubicin was provided to 56.6% and mitotane therapy to 53.3% of German patients with ACC (60 children, age 0.24–17.8 years) [16]. Redlich also concluded that mitotane treatment longer than 6 months and mitotane levels greater than 14 µg/ml in children were associated with significantly better survival [16].
Depending on the extent of the surgery and the result of histopathological examination (modified Weiss score/Wieneke index), patients with ACC are treated with adjuvant treatment (etoposide, adriamycin, cisplatin, and mitotane). Thus far, no international consensus has been reached on the best therapeutic approach to ACC beyond surgery in children.
ESMO-EURACAN clinical practice guidelines indicate that adjuvant mitotane is generally recommended in adult patients at high risk of recurrence, like stage III or R1-RX resection and/or Ki-67 index > 10%. However, adjuvant therapy should be discussed on an individual basis in patients with a low risk of recurrence (stage I/II, R0 resection, Ki-67 index < 10%) [11].
Chemotherapy, including mitotane in children, has been reported for advanced stages, but the standardized implementation of druggable targets has not been described yet.
Rodriguez-Galindo et al. published in 2021 the results of the clinical management of 77 eligible patients with stage I–IV ACC treated in 3 different interventional cohorts within the Children’s Oncology Group ARAR0332 trial [7].
According to clinical stage, patients were addressed to receive surgery alone (stage I), surgery and retroperitoneal lymph node dissection (RLND) (stage II), surgery plus RLND plus EDP chemotherapy for 8 cycles, and mitotane for 8 months (stage III–IV). The 5-year event-free survival (EFS) and overall survival by clinical stages were, respectively, 82.2 and 95.2% (stage I), 53.3 and 78.8% (stage II), 81 and 94.7% (stage III), and 7.1 and 15.6% (stage IV). In contrast with the ARAR0332 trial, according to adult international guidelines, mitotane therapy in locally advanced/metastatic ACC is continued until disease progression in cases of advanced disease and for at least 2 years in the adjuvant setting [11, 17, 18].
However, according to Grissanti, the mitotane treatment proposed by Rodriguez-Galindo for 8 months could be an undertreatment considering that approximately 4 months are required to attain a therapeutic range in the plasma, and approximately 10% and up to 40% of patients never reach the therapeutic range [19, 20].
The EXPeRT/PARTNER paediatric recommendation from 2021 suggests mitotane treatment for 2 years, during the time with the highest recurrence risk.
However, it should be noted that only periods with adequate therapeutic plasma levels should be considered effective treatment time [14].
In 2022, the results of the ADIUVO study were published. It is a randomized controlled study comparing the efficacy of adjuvant mitotane treatment vs. observation in prolonging recurrence-free survival (RSF) in adult patients at low-intermediate risk of recurrence adrenocortical carcinoma. A total of 91 patients were enrolled in ADIUVO, 45 in the mitotane and 46 in the observation arm. The main inclusion criteria were stage I-III ACC, R0 surgery, and Ki-67 ≤10%. The results of the ADIUVO study do not support the routine use of adjuvant mitotane in this subset of patients, who may thus avoid a potentially toxic treatment [21].
Clinical trials are underway testing the efficacy of monoclonal antibodies (pembrolizumab, nivolumab, ipilimumab) or multi-tyrosine kinase inhibitor (cabozantinib) in advanced ACC.
Weiss’s scoring system seems unreliable for paediatric ACTs, but later the Wineke index was reported to have a prognostic value more reliable than other histological prognostic scores used in adults [22]. This index classifies ACTs into 3 prognostic groups (benign, malignant, and of undetermined malignant potential) based on the number of present pathologic criteria (tumour size, tumour weight, extension into periadrenal soft tissues and/or adjacent organ, vena cava invasion, capsular invasion, necrosis, mitotic rate/atypical mitoses, and vascular invasion [≤ 2, benign; 3, undetermined malignant potential; ≥ 4, malignant]). More recently, Picard et al. described a 5-item microscopic score based on evaluating adrenal capsular invasion, venous invasion, tumour necrosis, mitosis >15/20 high-power fields, and Ki-67 index > 15%. ACTs with 2 or less features should be considered to show a “favourable histology”, and those with more than 2 features an “unfavourable histology” [23].
The Children’s Oncology Group Adrenocortical Tumours (COG ACT) Staging System for adrenal tumours contains stage I: R0 (complete histological resection) and small localized tumours (<100 gr or < 200 cm3) with normalization of hormone levels after surgery; stage II: R0 and large localized tumours (≥ 100 gr or ≥ 200 cm3) with normalization of hormone levels after surgery; stage III: Unresectable tumours or gross/macroscopic residual disease, tumour spillage, failure to normalize hormone levels after exclusive surgery, retroperitoneal lymph nodes involvement; and stage IV: distant metastases.
In our study group, 2 patients were treated with chemotherapy. In one of them, a year after the adrenalectomy and chemotherapy, metastasis in the collateral adrenal gland was observed, and it was treated with surgery, adjuvant chemotherapy, and mitotane. Mitotane was used in all patients, in one only in treating metastatic cancer.
Mitotane is an analogue of dichloro-diphenyl-trichloroethane (DDT) insecticide, which has been used for ACC since 1959. It causes necrosis of cells in all layers of the adrenal cortex. Mitotane has a long half-life, from 18 to 150 days; it is stored in tissues with a high lipid concentration and then released. It is known to cause frequent side effects, including endocrine disorders such as AI, hypothyroidism, and precocious puberty, central nervous system toxicity (lethargy, somnolence, dizziness, vertigo), gastro and hepatic disturbances (anorexia, nausea, vomiting, diarrhoea, hepatitis, or liver enzyme elevation), and increased serum cholesterol or leukopaenia. Mitotane in adults is initiated at a low dose (1 g/day with progressive weekly increases up to 2–3 g/day) or a high dose (3 g/day rapidly increasing to 6–9 g/day within 2 weeks) [24, 25]. Treatment should be initiated in children with doses 1.5 g/m2/day, then increased progressively to 4 g/m2/day in 2–3 doses. At a daily low dose of mitotane, the target plasma concentration is usually reached within 3–5 months [24]. Mitotane plasma levels in children should be monitored at frequent intervals (approximately every 2 weeks) until the therapeutic window has been reached; then they should be monitored every month. Mitotane is widely used in adults for adrenocortical carcinoma, and its efficacy for prolonging recurrence-free survival has been reported [26].
Adjuvant mitotane treatment in adults reduced postoperative recurrence by 38% and postoperative death by 31% [27]. There has been limited use of mitotane in children, and its usage remains to be established. So far, complications after treatment with mitotane have been described only in a few children who were treated with mitotane after surgery [28–30]. Monitoring the plasma level of mitotane is emphasized to exploit its therapeutic effects and avoid its toxic effects. Mitotane has a narrow therapeutic range of between 14 and 20 µg/ml [31]. Like in adults, a wide range of doses is used depending on the individual. Monitoring plasma levels of mitotane is necessary for effective and safe treatment, even after the determined dose. A mitotane level of more than 20 µg/ml is associated with neurological toxicity [31]. Endocrinopathies associated with use of mitotane include AI, PP, and hypothyroidism. Other adverse events are gastrointestinal, hepatic, and lipid disturbances or neurological symptoms.
Central/primary adrenal insufficiency
Mitotane has significant adrenolytic properties involving AI. Simultaneously with the start of mitotane treatment, glucocorticoid replacement therapy should be started; less often, mineralocorticoid treatment is necessary.
In vitro studies showed that mitotane causes interference with cholesterol metabolism and mitochondrial activity in adrenocortical cells, which often increases the apoptosis rate. It also reduces adrenal steroidogenesis by inhibiting key mitochondrial enzymes like CYP11B, which in turn reduces the conversion of hormone precursors into active hormones such as cortisol [32]. By comparing ACTH levels between patients treated with mitotane for ACC and patients with primary AI, it was also found that mitotane potentially interferes with pituitary function. Basal values and post-CRH stimulation test (100 µg) showed a statistically significant decrease in ACTH levels in patients treated with mitotane, suggesting an inhibitory effect at the hypothalamic-pituitary-adrenal axis [33]. In vivo studies revealed strong inhibition of 5∝-reductase activities and significant induction of hepatic CYP3A4/5 activities in mitotane-treated patients, with an obvious impact on the glucocorticoid and androgen replacement requirement during mitotane therapy [34, 35]. CYP3A4/5 are responsible for not only the conversion of cortisol to 6βOHF but also the 6β-hydroxylation of testosterone. The study conducted by Chortis, which analysed 24-h urine samples from 127 patients collected before and during mitotane treatment, demonstrated a 10- to 15-fold increase in 6βOHF/F excretion, establishing mitotane as one of the strongest inducers of CYP3A4 activity [34]. Mitotane increases the level of cortisol-binding globulin levels [36]. Both mechanisms mentioned above (hepatic enzyme induction and increased cortisol binding globulin [CBG]) suggested an increased dose requirement for glucocorticoid replacement in mitotane-induced AI.
In mitotane-treated patients, corticosteroids and ACTH values can be impaired by the interference of this drug. Serum cortisol cannot be considered as a valid parameter because mitotane increases CBG, which artificially raises total cortisol [37]. A more useful method can be represented by salivary cortisol (especially with tandem-mass spectrometry), which is unaffected by CBG alterations and reflects the serum-free cortisol status [38]. A recent study proposed the utility of hair cortisol measurement in mitotane-treated ACC patients for the long-term monitoring of hydrocortisone treatment [39].
Managing AI during mitotane treatment is a challenge. Chortis et al. suggest that glucocorticoid replacement in mitotane-treated patients should be initiated with at least double the dose normally used in primary AI, i.e. 40–50 mg hydrocortisone (equal to 75 mg cortisone acetate) rather than 20–25 mg hydrocortisone (equal to 37.5 mg cortisone acetate) per day [34].
In the Porier et al. study, 22 patients received glucocorticosteroids (GKS) (10-hydrocortisone, 10-cortisone acetate) during and after mitotane therapy. The mean maximal daily replacement dose was 69.4 mg (50–87.5 mg) for cortisone acetate and 51.6 mg (25–100 mg) for hydrocortisone during mitotane therapy. Mineralocorticoid replacement with fludrocortisone was necessary for 8 patients (34.8%) [40]. Additionally, after mitotane cessation, 18/23 (78.3%) patients achieved complete and 3/23 partial recovery of the hypothalamic-pituitary-adrenal (HPA) axis. The median time between the end of mitotane and GKS treatment was 1012 days (2.8 years) (min: 106 days, max: 2991 days) [40].
Dexamethasone should be avoided because it exerts a strong CYP3A4/5-inducing effect [41]. Oddie et al. described a 4-year-old girl after adrenalectomy due to ACC with adjuvant chemotherapy and mitotane treatment. As a result of adrenal insufficiency (AI), a hydrocortisone dose of 32 mg/m2/day was used [30]. According to other reports concerning children after surgical treatment due to ACC, followed by the use of mitotane, the dose of hydrocortisone was usually 30–50/m2/day [28]. LoGiurato et al. described a 2-year-old boy after surgical treatment for ACC. In this patient, the doses of hydrocortisone, up to 100–150 mg/m2/day, and fludrocortisone 0.5 mg/day, were used simultaneously with mitotane due to severe symptoms of AI. The change of the drug to prednisolone improved the patient’s condition [42].
In our study group, one patient presented symptoms of adrenal crisis after 5 months of mitotane treatment. Laboratory tests revealed hyponatraemia and hyperkalaemia. The dose of hydrocortisone was adjusted accordingly from 16 mg/m2/day to 40 mg/m2/day, and the treatment with fludrocortisone was introduced, improving the patient’s condition.
Mitotane and hydrocortisone (usually on a supraphysiological dose) were used simultaneously in all presented children. The doses of mitotane were gradually increased, obtaining a therapeutic concentration of the drug in the blood (14–20 µg/ml). The mean maximal dose of hydrocortisone was 21.72 mg/m2 (min: 17.4 mg/m2, max: 40.0 mg/m2). One patient received mineralocorticoid in a dose of up to 75 µg. The median duration of mitotane therapy was 10.5 months (min: 4 months, max: 55 months). The range of the initial dose of mitotane was from 0.5 g to 1 g, and the maximum dose was from 2.5 g to 6 g.
In 2 cases (1 and 2), no permanent adrenal damage occurred; HC was discontinued 7 and 12 months after mitotane cessation. Other patients still receive HC replacement therapy; however, in these cases, the time of observation after discontinuation of mitotane is short.
Central hypothyroidism
According to Poirier et al., mitotane therapy is frequently associated with the new onset of central hypothyroidism, with a prevalence of 95.5%; the study included 66 adult patients treated with mitotane for ACC [43]. Most of the cases occurred in the first year of treatment, 33.3% during the first 3 months of treatment, 3–6 months in 19.1%, 6–9 months in 14.3%, and 9–12 months in 9.5%; at least 14.3% of cases occurred after 12 months of exposure. Partial or full recovery of thyroid function occurred in 65.4% of cases, mainly in the first 2 years after mitotane discontinuation [43].
Another 4 studies reported hypothyroidism as an adverse event, identified by reduced FT3 serum levels, occurring in 68 of 141 patients whose thyroid function was measured [44–47]. What is the possible mechanism of hypothyreosis during mitotane treatment? Available data reported reduced total T4 (TT4) and FT4 serum levels, with no change in FT3 and TSH. These changes occurred after 3–6 months of treatment [44]. Mitotane can enhance the conversion of T4 to T3 by deiodinase, as confirmed by the FT3/FT4 ratio being in the upper reference range [48]. Moreover, mitotane could inactive TSH through abnormal glycosylation [49].
In vitro studies show that mitotane reduces the expression and secretion of TSH dose-dependently and blocks the TSH response to thyrotropin-releasing hormone (TRH), determining central functional hypothyroidism [50].
In our study, hypothyroidism was diagnosed in 2 patients. Test results showed decreased FT4 levels with normal FT3 levels and slightly elevated TSH concentration (Table II). The patients required temporary (up to 7 months) treatment with levothyroxine at the dose of 1.00 µg/kg/day and 1.13 µg/kg/day, respectively.
Table II
Test results and treatment before (I) and during mitotane therapy (II)
Peripheral/central precocious puberty
Peripheral precocious puberty (PPP) is the most common clinical presentation of ACC, seen in 50–80% of cases. Symptoms of hyperandrogenism in the form of pubarche praecox, acne, hirsutism, acceleration of growth, and advanced bone age are most often presented. Children with ACC may be at higher risk of central precocious puberty after treatment due to premature activation of the gonadotropin-releasing hormone pulse generator.
PPP may also be an adverse effect of mitotane use after surgical treatment of ACC, and then, due to advanced bone age, central precocious puberty (CPP) may be observed.
Mitotane has an oestrogenic effect that is oestrogen receptor α dependent (ER-α dependent) and mediated by mitotane-dependent increased hepatic sex hormone binding globulin (SHBG) synthesis [36]. The research conducted by Rossini et al. demonstrated that mitotane also directly binds ER-α as an agonist and suggested that selective oestrogen receptor modulators (SERM) or selective oestrogen receptor degrader (SERD) could be clinically useful to antagonize the oestrogen-like mitotane-induced effects [51]. Mitotane also has oestrogenic action, possibly impacting bone age advancement [30]. In the literature, there are only a few case reports of children in whom mitotane therapy recognized peripheral precocious puberty [28, 30]. To our knowledge, our report of 4 patients with PP during the mitotane treatment is the largest study.
Neurotoxicity
Other adverse effects observed in our patients included neurological complications.
All study group patients presented neurological symptoms of varying expression, which were more severe in younger children. The following manifestations were observed: dysarthria, unsteady gait, muscular hypotonia, decreased tendon reflexes, and limb tremors.
In the first patient, all neurological symptoms resolved after 6 months of mitotane treatment were discontinued. Complications were mainly connected with elevated serum drug concentration above the range.
Patient 2’s improvement in speech regression, dysarthria, stuttering, apathy, irritability, and excessive sleepiness occurred about 3–4 months after discontinuing mitotane. In patients 3 and 4, the symptoms of walk regression and lower limb pain regressed after about a month. Significant alleviation of limb tremors was also noted, occurring only with precise movements. Due to the short follow-up, we cannot determine the reversibility of symptoms in these 2 children.
Goto et al. described reversible encephalopathy (memory disturbance, spastic quadriplegia, and ataxia, mild cerebral and cerebellar atrophy in MRI, diffuse slowing of background activity in electroencephalogram [EEG]) in a 4-year-old boy 5 months after the start of mitotane treatment [29]. De Leon et al. reported a 15-year outcome in an infant with metastatic adrenocortical carcinoma treated with mitotane. The patient showed developmental delay and growth arrest [52].
A 5-year-old girl described by You Joung Heo developed cognitive and concentration disturbances with behavior regression 2 months after the start of mitotane treatment; at the last follow-up (at the age of 6.6 years), she exhibited only mild sleep and memory disturbances [28]. Based on the analysed literature, neurological symptoms usually, but not always, occurred in children with mitotane levels above the therapeutic level. Neurological symptoms were most often fully reversible after discontinuation of mitotane.
Gastrointestinal complications
Common side effects include lack of appetite, weight loss, nausea, vomiting, abdominal pain, and loose stools. Our patients presented similar symptoms. It is worth pointing out that common side effects of mitotane, such as gastrointestinal adverse effects, are difficult to differentiate from side effects of chemotherapy or signs of undertreated AI.
Hepatotoxicity
Both hepatocellular and cholestatic liver damage have been observed in patients treated with mitotane. Autoimmune hepatitis was also described [44]. It is probably due to direct hepatotoxicity. Mitotane is extensively metabolized in the liver via the microsomal enzyme system (predominantly 2C9) and producing a toxic or immunogenic intermediate can trigger liver injury. The severity of liver injury linked to mitotane therapy was generally mild and transient. Liver function tests should be periodically monitored, especially during the first months of treatment when or increasing the dose (Table II). In our study, alanine transaminase (ALT) was elevated in one patient, and aspartate transaminase (AST) concentrations were within the normal range during mitotane treatment. The gamma-glutamyl transpeptidase level was increased in 2 patients, in one of them significantly (up to 126 UI/ml). In one of our patients, 6 weeks after the start of mitotane treatment, cholelithiasis was confirmed using an abdominal ultrasound.
Lipid disturbances
Mitotane is a lipophilic agent that accumulates in circulating lipoprotein fraction and high-lipid-containing tissues. In adult patients during mitotane treatment, an increase in total cholesterol (TC), high-density lipoprotein (HDL), low-density lipoprotein (LDL), and triglyceride has been reported [44, 46, 53]. In the data of Vikner et al., a significant correlation was found between an absolute increase in HDL levels and mitotane concentration in plasma, but changes in total cholesterol, LDL, and triglycerides were not correlated with plasma mitotane [46].
In the study group, we observed slightly elevated TC levels in 2 patients (Table II). There are no data on lipid abnormalities during treatment with mitotane in the paediatric population. The mechanism of lipid disorders under mitotane treatment is still unclear. Two different mechanisms to explain the rise in lipids have been proposed: mitotane might stimulate the activity of 3-hydroxy-3-methylglutaryl-coenzyme A reductase, which is the rate-limiting enzyme in cholesterol synthesis, and thus, mitotane increases levels by inducing endogenous synthesis [54].
Alternatively, it has been suggested that mitotane inhibits CYP11A1, which would decrease the cholesterol conversion rate to pregnenolone [34]. Recently, mitotane was shown to confer adrenal-specific cytotoxicity and downregulate steroidogenesis by inhibiting sterol O-acyltransferase 1 (SOAT1), leading to lipid-induced ER stress [55]. Mitotane absorption also involves chylomicron binding, and mitotane binds to lipid membranes, modifying its physiochemical properties depending on the membrane lipid composition [56].
Conclusions
Adrenocortical carcinoma requires further investigation in children, especially concerning effective and safe adjuvant therapies. It is a matter of debate how long to use mitotane in children and how to deal with patients at high risk of disease recurrence who present with severe side effects regardless of the therapeutic concentration of mitotane in the serum.
It should be emphasized that the side effects associated with using mitotane should be recognized as soon as possible and adequately treated. The need to use supraphysiological doses of hydrocortisone, sometimes with mineralocorticoid, should be noted. Some patients require levothyroxine replacement therapy.
In the first patient’s case, mitotane treatment was maintained despite the increased symptoms of incomplete peripheral PP followed by central PP (GnRH analogue treatment was used and continued for 2.5 years). In 3 younger children, treatment was discontinued after one year, 8 months, and 4 months, respectively, due to rapidly progressing PP and neurotoxic symptoms.
The authors propose an algorithm of follow-up examinations in children treated with mitotane, considering possible side effects, i.e. PP, AI, hypothyroidism, neurotoxicity, and metabolic disorders (Table III).
Table III
Algorithm for endocrine safety, metabolic, and neurology assessment in children treated by mitotane
[i] TSH – thyroid-stimulating hormone; FT3 – free triiodothyronine, FT4 – free thyroxine; ACTH –adrenocorticotropic hormone; LH – luteinizing hormone; FSH – follicle-stimulating hormone; E2 – oestradiol; T – testosterone; GnRH – gonadotropin-releasing hormone; EEG – electroencephalogram; MRI – magnetic resonance imaging; ALT – alanine transaminase; AST – aspartate transaminase; GGTP – gammaglutamyl transpeptidase; TC – total cholesterol; HDL – high-density lipoprotein; LDL – low-density lipoprotein